
不同遗传背景自交系玉米根系微生物组差异比较
2022-05-25 02:23崔亚俊翟志文刘永鑫陈园园袁怀波严建兵
合肥工业大学学报(自然科学版) 2022年5期
崔亚俊,翟志文,王 超,刘永鑫,陈园园,袁怀波,严建兵,白 洋
(1.合肥工业大学 食品与生物工程学院,安徽 合肥 230601; 2.中国科学院遗传与发育生物学研究所 植物基因组学国家重点实验室,北京 100101; 3.中国科学院大学 生物互作卓越创新中心,北京 100049; 4.中国科学院遗传与发育生物学研究所 中国科学院-英国约翰英纳斯中心植物和微生物科学联合研究中心,北京 100101; 5.中国科学院大学 现代农业科学学院,北京 100049; 6.华中农业大学 作物遗传改良国家重点实验室,湖北 武汉 430070)
植物根系作为植物-微生物-土壤互作的主要部位,栖息着种类繁多的微生物群落,称之为根系微生物组[1],其中,细菌的定殖丰度最高,其他还包括真菌、卵菌、藻类、原生动物、线虫和病毒[2-4]。根系微生物中的有益微生物可以通过直接促进植物生长,或者帮助植物吸收水分、养分以适应逆境胁迫,如荧光假单胞菌[5]、菌根真菌[6]。因此,植物根系微生物组在植物生长发育、抗逆等过程中发挥重要作用[7],被称为植物第2个基因组[1]。近年来植物根系与微生物互作的研究逐渐增多,已是国际前沿热点[8]。
研究表明,植物微生物群落的系统发育结构由生物群落和环境因素相互作用所决定[9]。根系微生物群落结构由环境(如土壤、季节、地理位置、昼夜变化)、宿主(如基因型、生长阶段)、微生物-微生物和植物-微生物相互作用等因素共同决定[9-10]。在拟南芥、水稻等研究中均发现,根系微生物的组成由土壤类型决定,而宿主基因型在一定程度上决定根系微生物的比例[11-12]。在特定土壤类型及环境下,植物根系微生物组主要由植物根系分泌物和免疫代谢调控[13-14]。
玉米是世界三大粮食作物之一,不仅可以作为粮食、饲料生产以满足人类的基本生活需求,同时还是用来生产燃料乙醇的重要生物能源材料[15]。玉米在几千年的人工驯化过程中,产生了大量的遗传变异[16],而解析不同玉米自交系的遗传多样性是种质资源保护和育种的基础[17]。文献[18-19]通过对143个玉米自交系材料进行根系细菌群落组成研究发现,玉米根系细菌存在显著遗传力,数值在0.15~0.25之间,说明玉米基因型对其根系细菌丰度具有显著影响。
因此,本研究选取具有不同遗传背景的玉米自交系材料昌7-2和B73,在山东和海南试验田种植,通过对其根系样品16S rDNA特定片段的扩增及高通量测序,测定其根系微生物组成。通过根系微生物组群落组成、多样性及随机森林分析,揭示玉米基因型和栽培环境对玉米根系微生物组的影响,并找出可以显著响应玉米基因型的特征细菌类群,为玉米的遗传改良、解析遗传多样性提供理论基础。
1 材料与方法
1.1 试验材料与地点
试验选用不同遗传背景的优良自交系昌7-2、B73,分别于2017年11—12月在海南省三亚市华中农业大学南繁基地(109°10′E,18°21′N)和2018年5—6月在山东省东营市黄河口镇(118°50′E,37°41′N)进行种植。南繁基地试验点2017年11、12月平均气温分别为26.2、23.2 ℃,黄河口镇试验点2018年5、6月平均气温分别为20.3、25.2 ℃。
1.2 试验设计
南繁基地试验点于2017年11月1日播种,采用起垄播种,每个基因型玉米播种1行,行长2.5 m,株距25 cm、行距80 cm。播种前氮、磷、钾施用量均为100 kg/hm2,以复合肥方式施入。播种后30 d氮、磷、钾追施量分别为100、15、15 kg/hm2(m(复合肥)∶m(尿素)=1∶3)。
黄河口镇试验点播种日期为2018年5月1日,每个基因型玉米播种1行,行长2.5 m,株距25 cm、行距80 cm。氮、磷肥分别使用尿素75 kg/hm2和磷酸二铵20 kg/hm2。肥料均在播种前撒施,然后旋耕、覆膜、播种。在播种后27 d追施60 kg/hm2磷肥。
2个试验点均种植保护行。
1.3 测定项目与方法
1.3.1 样品采集
2个试验点的植株均长至八叶期时取样,海南和山东试验点取样时间分别为2017年12月6日和2018年6月12日。每次取样时,对于每个基因型,海南试验点分别选取3株、山东试验点选取6株有代表性的玉米植株,将根部用自来水冲洗,再用纯净水冲洗干净,用灭菌滤纸吸干水分,分别装在对应的15 mL无菌离心管内。样品全部冻存于冰箱。统一邮寄回北京实验室,-80 ℃保存。
1.3.2 扩增子文库构建及测序
在生物安全柜中将玉米根系样品剪碎,用样品研磨器(Precellys Evolution, Bertin Technologies, France)将样品研磨。利用土壤DNA提取试剂盒(FastDNA SPIN Kit, MP Biomedicals)提取根系样品总DNA。精确测定DNA质量浓度(PicoGreen dsDNA Assay Kit, Life Technologies,USA),随后稀释到3.5 ng/μL。利用引物799F和1192R对细菌16S rDNA的V5~V7可变区进行扩增,每个样本一式3份,聚合酶链式反应(polymerase chain reaction,PCR)反应体系为30 μL,含6 μL模板、0.75 U PrimeSTAR HS DNA聚合酶、1×PrimeSTAR缓冲液(TaKaRa, Japan),0.2 mmol/L dNTPs、10 pmol/L的带有barcode和连接序列的正向和反向引物及适量的无菌水。经过30 s 、98 ℃的初始变性步骤后,目标区域扩增25个循环,程序为98 ℃、10 s,55 ℃、15 s,72 ℃、60 s,最后在72 ℃延伸5 min。若阴性对照中没有出现扩增条带(没有添加模板),则将一式3份PCR产物混合,磁珠纯化(AMPure XP Kit,Beckman Coulter),使用Nanodrop(Nanodrop 2000c,Thermo Scientific)测定其质量浓度,并稀释至10 ng/μL,并作为第2轮PCR的模板。使用Illumina兼容性引物,对所有样本进行8个循环的重复扩增,PCR体系及程序与第1轮PCR相同。将每个样品的技术重复进行混合,使用胶回收试剂盒(QIAquick Gel Extraction Kit,Qiagen, USA),用1.2%琼脂糖凝胶进行切胶回收提取16S rRNA基因扩增产物。测定DNA质量浓度(PicoGreen dsDNA Assay Kit, Life technologies, USA),每个样品吸取200 ng并混合,然后磁珠纯化2次(Agencourt AMPure XP Kit,Beckman Coulter GmbH, USA),随后交于测序公司通过Hiseq-PE250测序平台进行高通量测序。
1.3.3 生物信息学分析
16S rRNA基因序列使用QIIME 1.9.1、USEARCH 10.0和内部脚本处理。双端序列使用FastQC过滤,并由usearch10 fastq-mergepair连接。Barcodes由usearch10-fastx-truncate提取。依据100%匹配阈值,由usearch10-unoise3挑选扩增子序列变异体(amplicon sequence variants,ASV)。以SILVA-132-SSURef-Nr99-tax-SILVA为参考数据库,用usearch10-uchime2-ref去除嵌合体,用USEARCH(usearch10-otutab)和自定义脚本去除非细菌序列。基于高置信度16S代表序列,由USEARCH生成ASV表。以RDP数据库为参考数据库,用usearch10-sintax对代表性序列进行分类,并用usearch10-sintax-u进行整理。
根据ASV表,使用usearch10-Alpha-div计算α多样性,并进行方差分析及Tukey HSD多重比较。在标准化ASV表的基础上,用beta-diversity.py计算β多样性。依据Bray-curtis距离计算得到的β多样性,利用R语言中vegan包进行限制性主坐标轴分析。为了获得可以显著区分2种基因型玉米根系微生物组的特征细菌种类,本文使用R语言程序包的默认参数对属级细菌的相对丰度与玉米基因型进行回归计算(R package ‘random Forest’,ntree=500,使用默认mtry=√p,其中p为属的分类单元数)。在R包“random Forest”中使用rfcv()函数,以实现5次交叉验证,从而确定重要特征细菌种类的数量和种类,并将其相对丰度经Z-score标准化处理,以热图形式呈现。
2 结果与分析
2.1 基因型和种植地对β多样性的影响
本文利用799F和1193R引物,以V5~V7区为扩增目标区域,通过16S rRNA基因扩增和Illumina测序获得每个根系样品(包括根表和根内)的细菌微生物组,总共从18个样本中测得1 902 310个高质量序列(平均52 842个,每个样本的序列数在32 791~86 781间波动)。本文使用Unoise去掉低丰度ASV(<8个总计数),通过对高质量序列进行分析,得到3 764个ASV。
通过对不同地点、不同基因型间根系微生物组的限制性主坐标分析发现,山东和海南试验点间玉米根系微生物组β多样性存在显著差异,同时每个试验点昌7-2和B73间也均存在显著差异,结果如图1所示,图1中不同颜色表示不同采样地点的不同基因型玉米根系微生物组的限制性主坐标分析(CPCoA),表明玉米根系微生物组β多样性同时受到种植地点和基因型的影响。进一步分析基因型和环境变异对根系微生物组β多样性的影响程度发现,两者都可以解释所有变异的35%,在限制性第1轴(83.47%)可以看出,海南和山东的样品明显分开,而在限制性第2轴(解释变异的16.53%上可以看出昌7-2和B73的样品在基因型之间明显分开。结果表明,相比于基因型,根系微生物组β多样性会在更大程度上受种植环境影响。
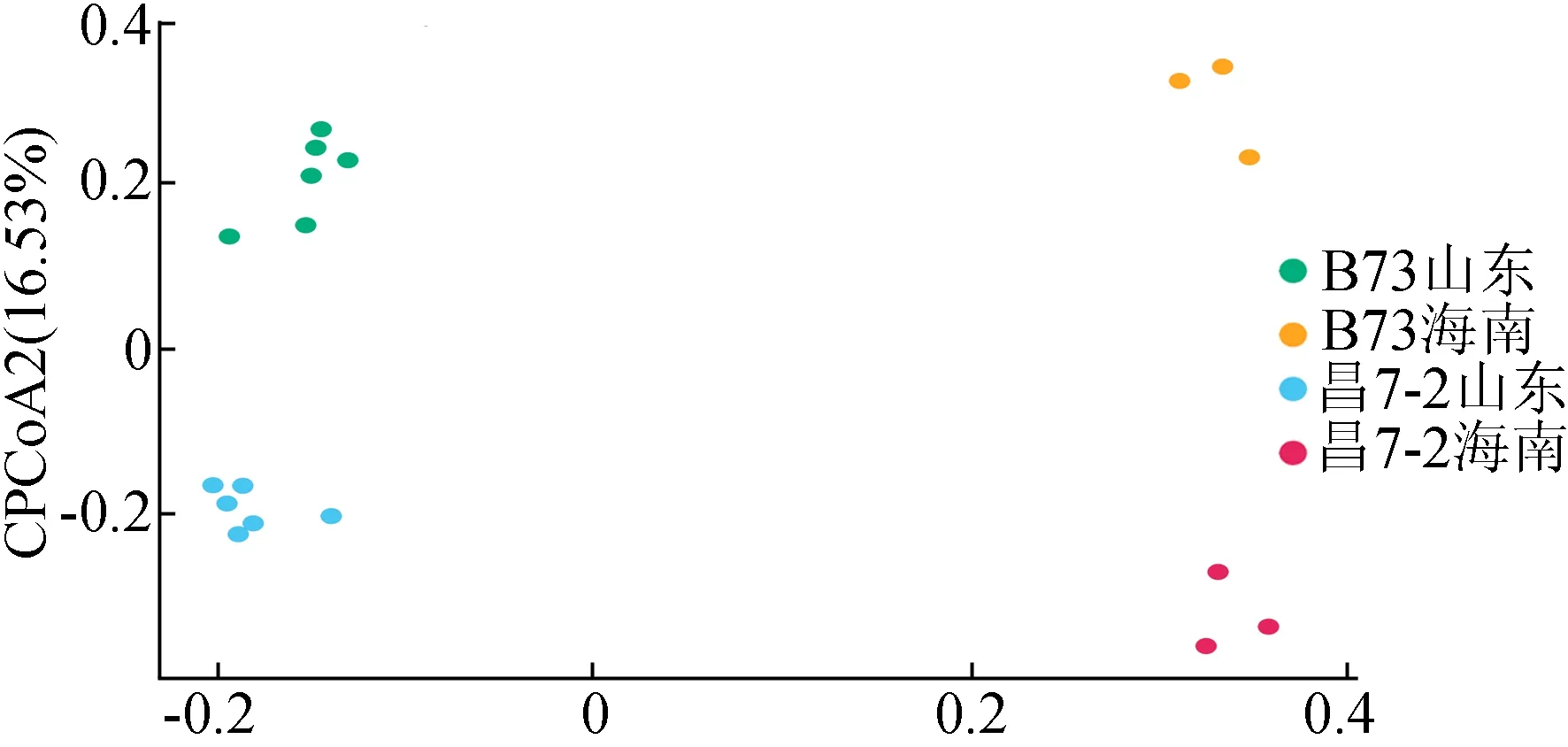
CPCoA1(83.47%)图1 玉米基因型昌7-2与B73间根系微生物组β多样性比较
2.2 基因型和种植地对组α多样性的影响
香农指数是将物种丰富度和物种丰度均匀度结合计算,反映生态系统物种多样性的一个经典指标[20]。通过对不同地点、不同基因型间根系微生物组香农指数的计算分析发现,同一试验点中,昌7-2和B73间物种多样性无显著差异,说明根系微生物组α多样性对基因型的响应较β多样性不敏感。昌7-2基因型香农指数在山东和海南种植点间存在显著差异,而B73在2个试验点间差异则未达到显著水平。种植地点对玉米根系α多样性的影响显著大于基因型对其影响,结果如图2所示,图2中,箱线图中横线代表样品对应数值的中位数;上、下边分别表示上四分位数和下四分位数;上、下竖线分别代表1.5倍的上、下四分位数;箱型图上标有相同小写字母表示组间差异未达到显著水平(P<0.05)。

图2 玉米基因型昌7-2与B73间根系微生物组香农指数比较
2.3 不同基因型和种植地根系微生物丰度比较
通过对玉米根系各种细菌相对丰度的计算发现,在2个种植地和2种基因型玉米的根系中,优势菌群均为Proteobacteria和Actinobacteria,2种细菌丰度平均占细菌总丰度的90%左右。2种玉米基因型根系中,Firmicutes在海南试验点玉米根系中的相对丰度显著大于山东试验点,说明海南试验点生态环境可能更有利于Firmicutes在玉米根系生长;昌7-2基因型玉米根系在山东和海南试验点间各门水平细菌相对丰度差异显著大于B73。另一方面,山东试验点中,昌7-2和B73基因型玉米根系中各门水平细菌相对丰度差异较小,而海南试验点则较大,具体表现为昌7-2基因型玉米根系富集更多Proteobacteria,而较少富集Actinobacteria,结果如图3a所示。
山东试验点中,玉米根系细菌优势菌属为Streptomyces、Lysobacter、Nocardioides、Sphingomonas、Ensifer和Bacillus,而在海南试验点中,玉米根系细菌优势菌属为Bacillus、Humibacter、Burkholderia和Streptomyces,反映了不同种植地点对玉米根系细菌种类的显著影响。山东试验点中,B73基因型玉米根系Streptomyces和Lysobacter相对丰度显著高于昌7-2,而在海南试验点中则是Humibacte和Streptomyces,结果如图3b所示。

图3 玉米基因型昌7-2与B73间定殖根系细菌相对丰度比较
2.4 基因型间存在显著定殖差异的细菌类群
为了进一步研究基因型间根系微生物组差异,排除栽培环境因素的影响,本文将山东和海南种植的2种基因型玉米共18个根系微生物样本合并分析。根据每个属在样品中的相对丰度中位数,选取相对丰度在0.5%以上的属为常见的共有属,这些属的相对丰度占全部细菌定殖丰度的79.21%。对这29个属利用随机森林分类器构建模型,以基因型为因变量,以属水平的细菌类群相对丰度为自变量。分析计算得出该分类器的错误率为22.22%,表明分类器的精确性较高。通过进行5次交叉验证表明,当利用29个属进行基因型间差异细菌类群的判别时,随机森林分类器的正确率最高。
根据平均降低精确性,本文选取29种菌中平均降低精确性大于0的细菌属作为判别2种基因型的特征属,共有18种,分别属于Rhizobiales(5种)、Actinomycetales(4种)、Xanthomonadales(3种)、Xanthomonadales(2种)以及Bacillales、Pseudomonadales、Sphingobacteriales、Sphingomonadales各1种,结果如图4a所示。

图4 昌7-2和B73玉米根系间具有显著差异的特征菌
图4a以基因型为因变量,以属水平的细菌类群相对丰度为自变量,应用随机森林分析判别方法确定了29个可以区分基因型的细菌种类,不同细菌种类对模型精度的重要性按降序排列;插图表示将输入的细菌种类数,按不同的重要性,与基因型进行回归得出的交叉验证错误率。具体属名及在不同基因型的玉米根系相对丰度热图如图4b所示,所有数据经Z-score标准化处理。所列举属水平细菌在玉米根系的丰度可能受到宿主遗传多样性的影响。
3 讨 论
3.1 栽培环境对微生物组的影响
植物根系作为植物-微生物-土壤互作的主要部位,既负责植株在土壤中的锚定、吸收水分和养分、物质储存,同时也生长有种类繁多的微生物,是植物与土壤微生物互作的主要部位[21]。本研究选取种植于不同种植地的不同基因型玉米,通过高通量测序,测定其根系细菌微生物组结构,通过限制性主坐标分析发现,不同种植地和不同基因型玉米根系细菌微生物组均存在显著差异,说明植物根系微生物组同时受到土壤环境和宿主基因型的影响。同时,相比于基因型,种植地可以解释更多的根系微生物组变异,说明玉米根系微生物组对栽培环境变化的响应程度大于基因型,这与以往的研究结果相一致[11-12]。目前认为植物根系从土壤中招募细菌的机制为两步选择假说。首先,根系分泌物和细胞壁促进了一部分土壤微生物的生长,从而导致根际细菌群落组成的变化和细菌数量的显著增多;然后,宿主与微生物的相互作用进一步调节根系内细菌群落的生长和比例[11,22]。因此,根系微生物的组成可能主要由土壤类型决定,而宿主基因型在一定程度上影响特定根系微生物的生长和定殖量,在拟南芥、水稻等研究中也发现类似结论[11-12]。
3.2 微生物组对栽培环境的响应
虽然生态环境是影响植物根系微生物组的主要因素,但在特定的土壤类型及环境下,植物基因型便成为影响土壤微生物群落结构的主要因素[23]。玉米在几千年的人工驯化过程中产生了大量的遗传变异[16],某些参与植物根系分泌物或免疫代谢的基因变异,会导致根系招募特定微生物的能力发生改变[11-14,24]。在本研究结果中,玉米根系细菌微生物组在不同基因型间存在显著差异,说明来自不同遗传背景的玉米植株在招募根系微生物方面也存在遗传多样性,与文献[19]的研究结果一致。本文通过分析香农指数和各门水平细菌相对丰度在山东和海南试验点间的差异,进一步发现,昌7-2均显著大于B73,说明昌7-2更易受到环境影响,而B73根系微生物组遗传力可能更高。以往研究表明,植物免疫代谢、根系分泌物以及磷饥饿响应代谢参与植物根系微生物组的组装,文献[25]发现拟南芥磷饥饿响应(phosphate starvation respones,PSR)相关基因参与根际微生物群落的正常“组建”,其中关键因子PHR1同时调控PSR和免疫响应。缺铁胁迫下,拟南芥根部特异性转录因子MYB72可促进以香豆素东莨菪亭为主的根系分泌物,进而影响根系有害微生物的生长[26]。昌7-2和B73根系微生物组存在显著差异且受栽培环境改变的影响程度不同,可能与其在以上几个生理代谢途径的差异有关。
3.3 宿主对细菌类群丰度的影响
玉米根系微生物组主要由Proteobacteria等组成[18-19]。有研究表明,根际细菌在植物根系的定殖变化与其自身特定生理代谢活性有关,如氨基酸、有机酸转运蛋白和糖苷水解酶等[27]。本文利用根系细菌的相对丰度,采用随机森林模型,找到若干与玉米基因型显著相关的特征细菌种类。结果表明,基因型辨识度较高的属为Rhizobium、Sphingobacterium等。另外,Streptomyces在图3b中也显示出基因型间显著差异,说明这些根系细菌相对丰度受宿主基因型影响的程度较高。许多放线菌可以产生抗生素,从而帮助植物抑制病原菌生长[28-29]。同时,Rhizobium和Sphingobacterium对宿主的促生作用也多有报道,如固氮、分解根系有毒物质等[30-31]。以上结果表明,受植物宿主基因型显著调控的根系细菌,可能在宿主响应环境胁迫过程中具有明显促生作用,从而显著受到宿主的主动调控,这可能是植物与微生物长期协同进化的结果[27-32]。
4 结 论
本研究选取种植于不同地点、具有不同遗传背景的玉米自交系材料昌7-2和B73,并对其根系微生物组进行高通量测序。研究结果表明,玉米根系微生物组对栽培环境变化的响应程度大于基因型,而不同基因型玉米根系微生物组对栽培环境变化的响应程度也不同,同时利用随机森林分析发现若干丰度显著受宿主基因型影响的细菌类群。
猜你喜欢
传染病信息(2022年4期)2022-11-23
现代园艺(2022年7期)2022-11-19
南京师大学报(自然科学版)(2022年2期)2022-06-20
安徽农学通报(2022年6期)2022-04-07
湖北农业科学(2019年22期)2019-12-23
农家致富顾问·下半月(2019年7期)2019-09-28
新课程·下旬(2018年8期)2018-11-10
大众科学(2018年1期)2018-03-28
中学生理科应试(2016年4期)2016-11-19
小星星·阅读100分(高年级)(2015年11期)2015-11-28
