
基于GBS-seq的青藏扁蓿豆SNP标记开发
2022-06-09 08:48周晓楠徐金青雷雨晴王海庆
生物技术通报 2022年4期
周晓楠 徐金青 雷雨晴 王海庆
(1.中国科学院西北高原生物研究所 中国科学院高原生物适应与进化重点实验室,西宁810001;2.中国科学院大学,北京100049;3.青海省作物分子育种重点实验室,西宁810001)
青藏扁蓿豆(Medicago archiducis-nicolai)是豆科(Fabaceae)苜蓿属(Medicago)多年生异花授粉二倍体植物[1],分布于青藏高原及其毗邻的高海拔高寒地区,对寒冷、干旱等极端环境具有极强的适应性[2-3],是青藏高原天然高寒草地上具有驯化潜力的野生牧草种质资源。遗传多样性是种内个体间或一个群体内不同个体遗传变异的总和,研究物种的遗传多样性不仅可揭示该物种群体间的遗传结构、进化关系,以及与环境及地理分布之间的相关性[4],也可以了解物种的遗传背景及亲缘关系,对种质资源的评价利用具有重要意义。
目前,核基因组(ITS[3])、叶绿体基因组(psbA-trnH[3]、trnL-trnF[5])标记以及简单重复序列(EST-SSR[6-7])等分子标记技术已被应用于青藏扁蓿豆群体遗传多样性研究。利用ITS和psbA-trnH遗传标记对不同海拔的青藏扁蓿豆野生群体进行遗传分析,发现遗传分化程度和采样点海拔高度差之间存在显著的相关性,表明海拔高度引起的遗传隔离是产生群体间遗传分化的主要原因。基于EST-SSR标记进行的遗传多样性分析,发现青藏扁蓿豆群体间遗传距离与地理距离和海拔差异之间均存在极显著的相关性,同时也提示由海拔差异导致的局部气候环境的异质性可能对青藏扁蓿豆野生群体的遗传多样性产生影响,并使之获得对局部极端生存环境的适应。但上述分子标记技术密度较低、多态性较低且操作复杂,具有一定的局限性。因此,为了进一步解析青藏扁蓿豆对极端环境的适应机制,评价青藏扁蓿豆种质资源,需要对青藏扁蓿豆开发更高效的分子标记类型。
单核苷酸多态性标记(single nucleotide polymorphism,SNP)作为第三代分子标记,已在群体选择分析[8]、重要性状基因定位[9]、人类群体扩张研究[10]以及动植物遗传连锁图谱构建[11]等相关研究中广泛应用,是研究物种遗传变异的理想分子标记[12]。与以SSR为代表的二代分子标记相比,具有易于实现自动化分析、多态性高及密度高等优点。但SNP标记由于前期测序阶段的高成本,限制了其大规模的开发利用,GBS(genotyping-by-sequencing)是基于二代测序技术分型SNP的一种简化基因组测序技术,是一种高效、简单、低成本的基因分型方法,对了解种质资源的遗传背景和系统进化,研究群体遗传多样性及遗传结构具有重要意义[13]。
本研究以5个群体共80份野生青藏扁蓿豆为材料,采用GBS技术测序,进而开发SNP标记,基于开发的SNP标记,初步进行遗传多样性及遗传结构分析,为后续更加全面解析青藏扁蓿豆在青藏高原极端环境下的适应机制提供数据支持,同时也为青藏扁蓿豆种质资源评价奠定基础。
1 材料与方法
1.1 材料
80份供试材料于2017年采集自青海省(表1),采样群体海拔分布于2 462-3 311 m,每个群体采集16个个体,采集的单个个体放入信封袋用硅胶干燥保存备用。

表1 青藏扁蓿豆材料来源Table 1 Sources of M.archiducis-nicolai materials
1.2 方法
1.2.1 基因组DNA的提取及纯化 取约0.05 g干叶用组织研磨仪进行研磨,利用改良的CTAB法[14]提取总DNA,提取缓冲液中加入少量的聚乙烯吡咯烷酮(PVP)粉末和β-巯基乙醇(14.4 mol/L),水合时间延长至5 h,期间每隔10 min震荡混匀一次,使干叶细胞充分裂解,异丙醇沉淀时间延长至90 min,提高DNA产率。DNA产物经0.8%琼脂糖凝胶电泳检测,确认目的条带清晰明亮且完整后,利用TIANGEN Purification Kit柱式胶回收试剂盒(增强型)进行纯化,纯化后的产物经过浓度及纯度检测达到测序要求后,-20℃保存备用。
1.2.2 GBS测序文库的构建 提取的DNA样品送往北京奥维森基因科技有限公司,参照Elshire等[15]建库技术(取样-酶切-接头连接-混样-PCR扩增-纯化)建库,首先应用限制性内切酶ApeKⅠ对基因组DNA进行酶切,加上带有条形码的接头后,对每个样品进行扩增,然后对样品进行混合,电泳回收375-400 bp区间的DNA条带,纯化后产物用于测序,测序反应在Illumina HiSeqPE150测序平台上进行双末端150 bp的测序。
1.2.3 SNP开发 将测序得到的原始数据按照如下标准过滤:剔除带接头(adapter)的reads pair;单端read中含有N的含量超过该条read长度比例的10%时,去除此对paired reads;单端read中含有的低质量(质量值Q≤5)碱基数超过该条read长度比例的50%时,也需去除此对paired reads。最终得到Clean reads进行后续分析。因为青藏扁蓿豆尚未进行全基因组测序,所以首先选取碱基数量(176 814 300)最多的 HY_14基于 Stacks2.4[16]无参分析的流程构建拟参考基因组,参数为-m=3,-M=2,-N=2;然后通过短序列比对软件BWA(http://bio-bwa.sourceforge.net/)将每个样本过滤后的高质量序列与拟参考基因组比对,参数:MEM-t=4,-k=32,-M;最后根据比对结果,利用GATK3.8(https://software.broadinstitute.org/gatk/)开发 SNP位点并使用Vcftools-v0.1.13(http://vcftools.sourceforge.net/)软件对开发的SNP位点进行以下条件的过滤:对任意一个位点,群体里至少有80%的样本有基因型、质量值最低为20、次等位基因频率最小为0.05并且次等位基因数量大于3,从而获得高质量的SNP位点。使用VCFTOOLS软件对SNP分型结果进行转换(transition)与颠换(transversion)统计并由EXCEL绘图。
1.2.4 遗传数据分析 利用ADMIXTURE(http://software.genetics.ucla.edu/admixture/download.html)软件检测群体的遗传结构,所得结果通过Excel进行可视化绘图;采用GCTA(https://cnsgenomics.com/software/gcta/bin/gcta_1.93.2beta.zip)进行主成分分析(principle component analysis,PCA),并使用R 包 ggplot2[17]画图 ;利用 ARLEQUIN 3.5[18]软件计算遗传多样性指数(Pi)、观测和期望杂合度(HO和HE)以及遗传分化指数(FST),进行遗传多样性和遗传分化程度分析;利用GENALEX6.503[19]软件中的Mantel test对成对的群体统计地理距离,并计算其与遗传距离的相关性。
2 结果
2.1 测序质量
对80份青藏扁蓿豆材料的测序数据进行统计,包括5个群体的平均测序片段数,平均碱基数,群体Q30与GC含量,以及酶捕获率。结果(表2)显示,测序共获得60.79 Gb数据,平均每个样本0.76 Gb,5个群体产生的测序片段数以及碱基数存在差异,其中,最多的为湟源(HY)群体(平均测序片段数为6 638 025条,平均碱基数为0.99 Gb),最少为西宁西山(XNXS)群体(平均测序片段数为4 360 916条,平均碱基数为0.65 Gb),平均Q30≥93.07%,平均GC含量≥42.29%,数据满足分析要求。经过低质量数据过滤后得到的平均测序片段数为3 468 857(XNXS)-5 456 407(HY),群体测序平均酶捕获率≥98.74%,总计保留下12 796个高质量SNP位点用于后续分析。

表2 青藏扁蓿豆群体测序数据统计Table 2 Summary of sequencing data of M.archiducis-nicolai populations
2.2 SNP突变类型及数量
对SNP分型结果进行突变类型统计,结果(图1)表明,C/T转换类型最多(4 124个),占所有碱基突变类型的32.23%,A/G转换类型(4 037个)占31.55%;G/C颠换类型最少(518个),占所有碱基突变类型的4.05%,A/C(1 164个)、A/T(1 777个)和G/T(1 176个)颠换类型分别占9.10%、13.89%和9.19%。转换与颠换(Ts/Tv)之比为1.761。
2.3 遗传结构分析
基于SNP分型数据,对来自5个群体的80份青藏扁蓿豆材料进行群体遗传结构分析,提取K=2-5时的交叉验证错误率(cross-validation error,CV error)(图2-A),可见K从2-5交叉验证错误率值逐渐增大,因此,可将K=2作为最佳K值。当K=2时,群体遗传结构图将青海祁连(QLCD)群体与其他群体划分开(图2-C)。主成分分析以得到的SNP为基础进行分析(图2-B),结果显示,个体的聚类情况能够清楚地被反映出来,亲缘关系的远近也可以由群体之间的距离反映。第一主成分(PC1)将青海祁连(QLCD)群体与其他群体区分开,这与ADMIXTURE分析的结果相一致。
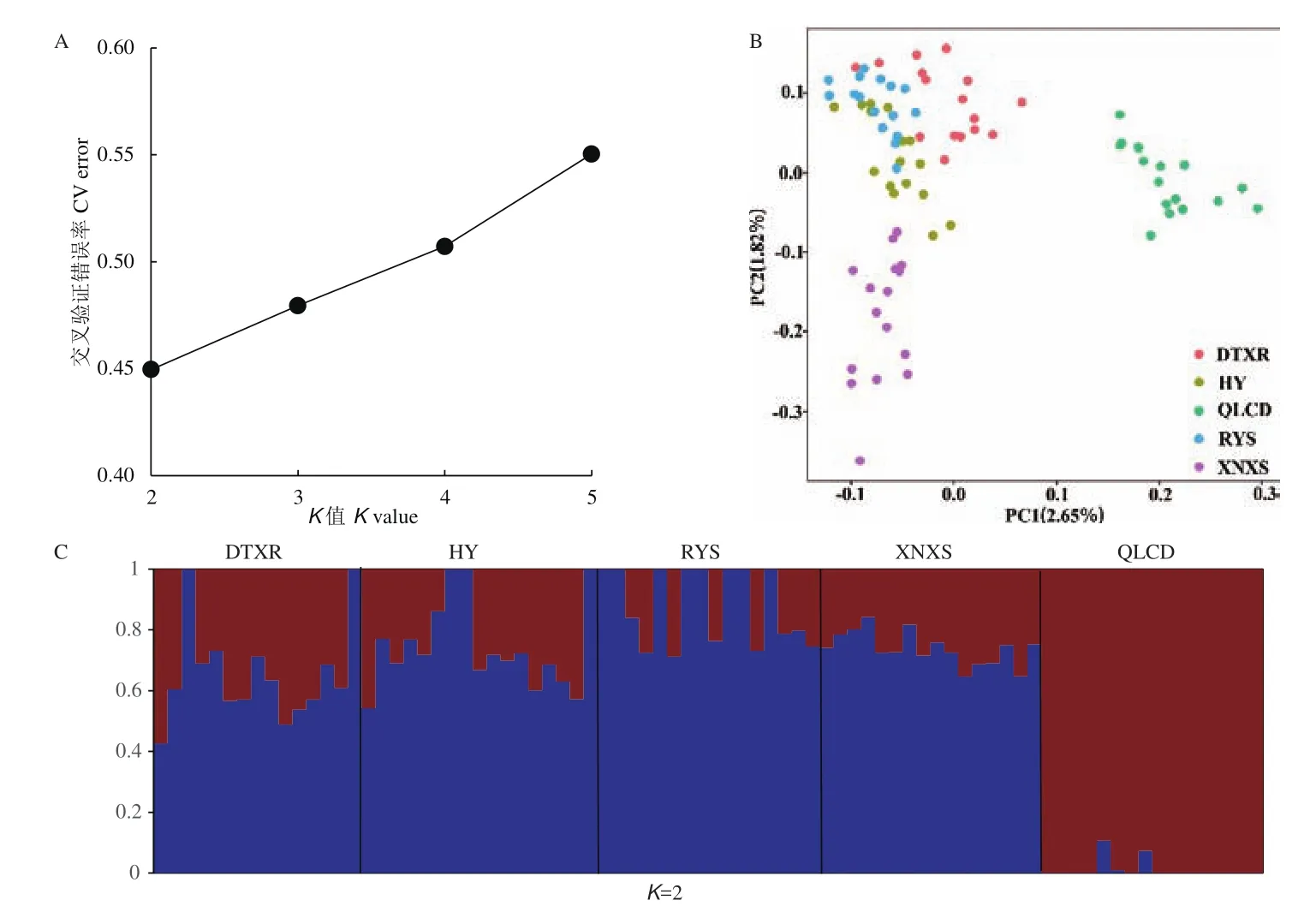
图2 群体遗传结构分析Fig.2 Population genetic structure of populations
利用遗传分化指数(FST)评估青藏扁蓿豆群体间的遗传分化程度,发现群体间的遗传分化指数为0.009 3-0.036 9,所有群体间均存在极显著(P<0.01)的遗传分化(表3)。基于Mantel test分析,结果表明,青藏扁蓿豆野生群体的地理距离与遗传距离之间存在极显著的正相关关系(R2=0.877 6,P=0.006,图3)。
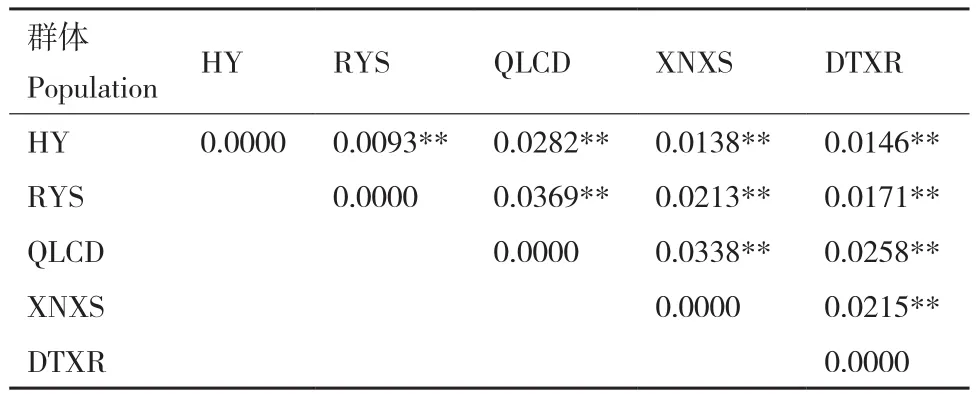
表3 群体间遗传分化指数(FST)Table 3 Index of genetic differentiation(FST)between populations
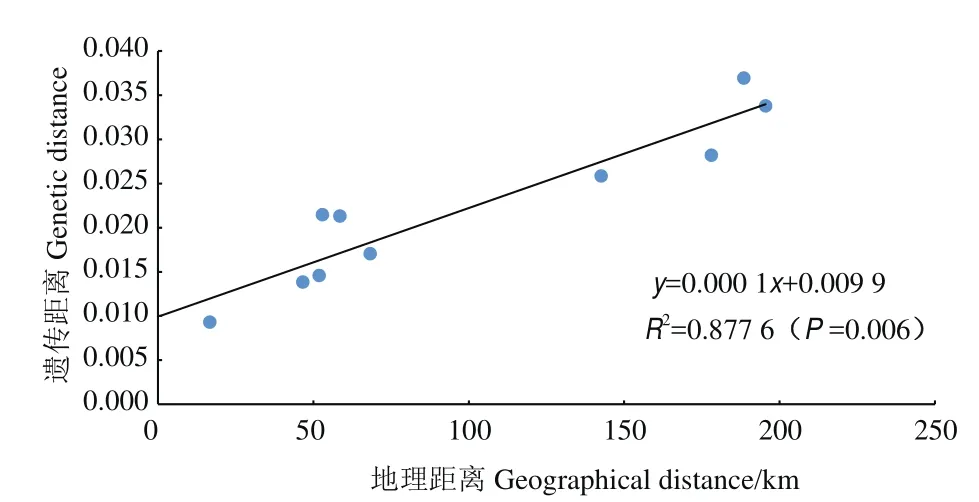
图3 群体遗传距离与地理距离的相关性Fig.3 Correlation between population genetic distance and geographical distance
2.4 遗传多样性分析
基于开发的12 796个SNP位点进行遗传多样性分析,发现各群体的平均等位基因数(NA)均为2;观测杂合度(HO)在0.187 68(RYS)-0.304 36(QLCD);期望杂合度(HE)在0.201 97(RYS)-0.364 34(QLCD);遗传多样性指数(Pi)为0.178 32(RYS)-0.241 34(QLCD)。 表 明 日 月 山(RYS)群体具有相对最低的遗传多样性水平,青海祁连(QLCD)群体具有相对最高的遗传多样性水平(表4)。
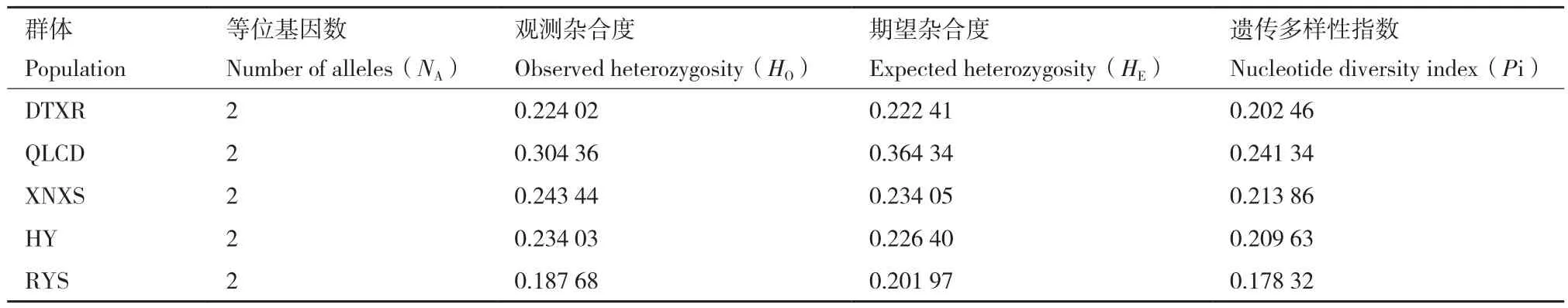
表4 青藏扁蓿豆野生群体遗传参数统计Table 4 Statistics of genetic parameters in M.archiducis-nicolai wild populations
3 讨论
简化基因组测序技术适合于大样本量的研究,可快速鉴定出高密度的变异,特别是SNPs变异,在物种进化、遗传多样性分析、全基因组关联分析等领域中应用越来越广泛。其中,GBS技术运用了甲基化敏感的限制性内切酶,回避了基因组主要的重复区域,使得文库构建简单,成本相对低廉,且有助于高通量SNPs分型技术的大规模应用。前人对烟草进行GBS测序,利用得到的SNP位点建立的聚类树将供试材料进行了准确划分[20]。也有研究表明,利用GBS技术对紫花苜蓿品系进行测序,并结合表型性状进行全基因组关联分析,可得到与黄萎病性状显著相关的SNP标记[21]。本研究利用GBS技术对80个青藏扁蓿豆样本进行测序,得到了60.79 Gb的原始数据,经过挖掘并过滤最终获得了12 796个高质量的SNP位点,用于后续遗传多样性和遗传结构分析。
碱基置换突变是形成生物个体多态性和推动种群进化的根本原因之一,大部分碱基置换突变只涉及单个碱基的替换,即转换(transition)和颠换(transversion)[22]。转换是嘧啶互换或嘌呤互换,如C/T和A/G型SNP;颠换则是嘧啶和嘌呤相互替换,如A/C、C/G、A/T和G/T型SNP。本研究中所获得的SNP位点碱基转换类型占63.78%,远高于碱基颠换(36.22%)的占比,与严佳文等[23]研究的火龙果碱基变异类型(转换占63.29%,颠换占36.71%)相同。由于转换有4种可能性,颠换有8种可能性,因此,理论上发生转换概率与发生颠换概率的比值(Ts/Tv)应该等于0.5。但实际上,Ts/Tv值往往大于0.5,这种差异性被称为“转换偏差”[24-26]。本研究挖掘的SNP突变类型存在明显的转换型偏差现象,Ts/Tv=1.761>0.5,产生这种差异的原因可能与不同物种在进化中承受的选择压力有关[27]。
一个物种的进化潜力和抵御不良环境的能力既取决于种内遗传变异的高低[28],也有赖于遗传变异分布格局,即群体的遗传结构[29-30]。本研究对青藏扁蓿豆野生群体的群体遗传结构分析表明,5个青藏扁蓿豆群体划分为2个大的类群,而之前研究并没有将其分开[7],因此,SNP标记与EST-SSR标记相比具有更高的分辨率。群体的遗传分化指数(FST)是衡量群体间遗传分化程度的重要参数,可以解释影响种群发生遗传分化的因素[31]。本研究5个群体中,群体分化指数为0.009 3-0.036 9,且所有群体间的群体分化指数均小于0.05,根据Wright[32]对遗传分化指数的界定,FST<0.05表明群体遗传分化较弱,0.05<FST<0.15表明群体中等程度遗传分化,0.15<FST<0.25表明群体较大程度遗传分化,FST>0.25表明群体极大程度遗传分化。因此,本研究中群体间遗传分化较弱。此前研究显示,来自青海卓尼、青海乐都以及内蒙古的扁蓿豆供试材料的群体分化指数为0.047 1-0.138 2,青藏扁蓿豆供试材料中的群体分化指数介于-0.055 2-0.062 4[5],表明青藏扁蓿豆群体的传分化程度较低。本研究中的5个青藏扁蓿豆群体间的分化指数也较低,表明各群体间遗传分化程度均较弱、亲缘关系相对均较近。对青藏扁蓿豆群体遗传距离与地理距离的相关性分析表明,地理距离与群体的遗传分化存在极显著(P<0.01)的相关性,与此前基于EST-SSR标记研究的结果相一致[7],推测可能与冰期后居群由避难所向研究群体扩散过程中发生的奠基者效应有关。
高原抬升,亚洲季风、亚洲内陆干旱化以及第四纪冰期等诸多因素共同作用造就了青藏高原复杂独特的地貌环境和丰富的遗传多样性[33]。本研究发现,位于青藏高原北部的青海祁连(QLCD)群体具有相对较高的遗传多样性,而此前研究表明,来源于青藏高原东南边缘的青藏扁蓿豆群体具有相对较高的遗传多样性[6-7],推测高原东南边缘可能存在青藏扁蓿豆冰期避难所。造成这种结果的差异可能是由于采样群体数过少,且群体分布相对集中,依据并不充分。因此,下一步可扩大采样群体范围来进行后续研究分析。
4 结论
运用GBS测序技术在80份野生青藏扁蓿豆材料中开发了12 796个高质量的SNPs。青海祁连(QLCD)群体的遗传多样性水平相对最高;5个群体被划分为2个大的类群,此结果与主成分分析结果基本一致。另外,该青藏扁蓿豆群体间的遗传分化程度较弱,地理距离与群体的遗传分化程度呈现极显著(P<0.01)的相关性。
猜你喜欢
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
教学考试(高考生物)(2020年6期)2020-11-23
阿来研究(2020年1期)2020-10-28
中西医结合肝病杂志(2020年2期)2020-10-27
文苑(2020年6期)2020-06-22
食品与生物技术学报(2020年8期)2020-01-06
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
福建基础教育研究(2019年4期)2019-05-28
