
基于Cheng-范数的不同维度样本相似性度量
2022-07-04 03:20郭志伟陈新庄
延安大学学报(自然科学版) 2022年2期
郭志伟,陈新庄
(延安大学数学与计算机科学学院,陕西延安 716000)
生物信息学中,为解决蛋白质功能聚类、疾病相关基因识别及药物发现等问题,学者们已经从各个计算方面提出了许多强大的机器学习方法,如决策树、支持向量机、朴素贝叶斯及深度学习方法等[1-3],样本相似性度量在这些方法中均起着至关重要的作用。可是,这些实际问题涉及的样本,如蛋白质、基因及药物等,通常具有不同维度。然而,大量现有经典相似性度量,如曼哈顿距离、欧几里得距离、切比雪夫距离及余弦距离等,均仅适用于相同维度样本。因此,无法通过经典相似性度量方法来度量不同维度样本相似性。
为了克服这些困难,SAW 等[4]提出了一种无对齐方法,从样本序列角度来度量不同维度样本相似性。无对齐方法通常分两步来度量样本序列相似性:第一步,将样本序列转换为固定长度的特征向量;第二步,以这些提取的特征向量相似性来衡量原始样本序列相似性。在这个过程中,特征提取对无对齐方法的准确性起着关键性的作用,但是从原始数据提取有效特征并非容易实现。目前,使用描述符来获取固定长度特征向量是一种有效方法。FAN 等[5]通过组合多种已有描述符提出了一种有效描述符,来度量蛋白质序列相似性。可是,描述符的设计或定义通常不是一件容易的事,而且容易丢失一些不确定是否有用的信息[6-8]。
同时,也存在一些方法从样本三维结构角度来度量不同维度样本相似性。样本三维结构通常比样本序列包含更多信息,样本三维结构由构成其分子的原子空间分布决定。因此,可通过原子空间分布相似性来衡量样本三维结构相似性。ANKERST等[9]将蛋白质空间均匀划分,并生成每个区域的统计信息,从而将蛋白质的三维结构转化为形状直方图。HU 等[10]根据蛋白质分子结构的分形特征,提出了描述蛋白质三维结构的体积分形参数。AKUTSU[11]根据所比较蛋白质对的骨架在空间中可以对齐的程度,衡量了两者的相似性,在此过程中,蛋白质结构的骨架坐标被严格平移和旋转,并利用RMSD 方法能使其整体结构在另一蛋白质骨架上最大重叠。这些方法的关键是提取样本结构中空间旋转和平移不变的特征。这个特征量本身可以是几何、拓扑或其他与空间位置无关的生物信息。但是,无论从哪个方面提取特征,都有可能丢失一些关键信息。为了克服这些不足,CHEN 等[12]最近通过将样本三维结构转化为图,并引入新的矩阵克罗内克积(p,m)-范数,从而通过图核、矩阵克罗内克积(p,m)-范数来度量药物之间的相似性,进行药物发现。
本文在CHEN 等[12]近期工作的基础上,基于由著名非线性系统控制理论专家程代展研究员引入的Cheng-范数,提出一种不同维度样本相似性度量方法。该方法的基本思路是:将样本三维结构转化为图,并给出图对应的二维矩阵表示,通过基于Cheng-范数定义的矩阵相似度来度量样本相似性。同时,以蛋白质功能聚类为例,通过该样本相似性度量方法,分别基于蛋白质加权图对应的加权邻接矩阵和加权拉普拉斯矩阵对蛋白质样本进行相似性度量,从而采用层次聚类算法对其进行功能聚类,以验证该相似性度量方法的有效性。该方法采用了图表示,能够保留样本的结构信息,而且相比于矩阵克罗内克积(p,m)-范数,矩阵Cheng-范数更容易被计算。
1 基本符号与术语
在介绍基本术语之前,首先给出本文中经常用到的一些符号:
1)[m,n]表示正整数m 与n 的最小公倍数,并用(m,n)表示正整数m与n的最大公因数;
2)⊗表示矩阵的克罗内克(Kronecker)积(也称张量积);
3)1n表示n维全1列向量(1,1,…,1)T;
4)In表示n阶单位方阵;5)(x,y)表示向量x和y的数量积(也称内积)。接下来,介绍由程代展研究员定义的一些基本术语[13-15]。
1.1 Cheng-MV积

1.2 Cheng-内积与Cheng-范数

设x是m维实向量,则x的Cheng-范数定义为

2 基于Cheng-范数的相似性度量方法
本文所呈现的样本相似性度量方法的基本思路是:将样本三维结构转化为图,并给出图对应的二维矩阵表示,通过基于Cheng-范数定义的矩阵相似度来度量样本相似性。
为了定义矩阵相似度,通过将不同阶数方阵映射成相同阶数方阵的思想,基于Cheng-范数,首先引入不同阶数方阵的距离。
定义1 设A 和B 分别是阶数为n 和m 的实方阵,则A和B的距离定义为

该样本相似性度量方法通过不同阶数方阵的相似度,来度量相应不同维度样本的相似性。
3 蛋白质功能聚类
在蛋白质功能预测中,比如想预测给定蛋白质是否是一种酶,通常采用的方法是通过寻找具有相似序列、结构或化学性质的蛋白质来推断给定蛋白质功能。一种非常有效的方法是将蛋白质的三维结构转化为其对应的表示图(如图1 所示),并通过图的相似性来度量蛋白质的相似性[16]。
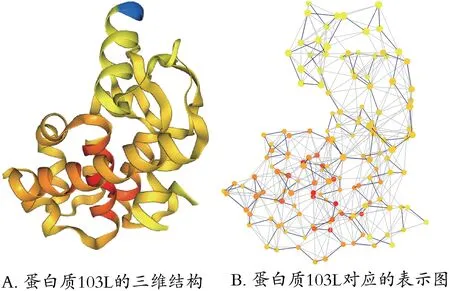
图1 蛋白质103L的三维结构及表示图
本实验从蛋白质数据库(PDB)[17]选取60 个蛋白质样本,其中30 个为O-糖基水解酶,以XXXL 格式命名,即103L、104L;其余30 个为氧转运蛋白,主要分为两部分(也有个别例外情形,如1YOG):一部分以XXXM 格式命名,即105M、107M,另一部分以2ZXX 格式命名,即2ZSN、2ZT0。进一步,将蛋白质样本转化为图,并给出其矩阵表示。通过定义的不同阶数方阵相似度,来度量相应不同维度样本相似性,并对选取的60 个蛋白质样本进行功能聚类,以聚类结果来验证该相似性度量方法的有效性。
3.1 蛋白质结构的二维矩阵表示
本实验从蛋白质结构图分析(NAPS)[18]获取这60个蛋白质的加权图[19]。在蛋白质加权图中,由Cα原子表示的氨基酸残基被视为图的节点;若一对Cα原子之间的距离在上、下阈值范围内(0~7Å),则在这对节点之间连一条边;定义边权重为所连接Cα原子对欧氏距离的倒数。图1B 展示了蛋白质103L 对应的图。通过蛋白质结构图分析(NAPS),可获得这60 个蛋白质加权图的全局参数信息。在下列实验中,首先以这60个蛋白质加权图的加权邻接矩阵作为其二维矩阵表示,来度量相应不同维度样本相似性,并进行功能聚类。
3.2 蛋白质功能层次聚类
不难看到,不同维度蛋白质的加权图对应着不同阶数加权邻接矩阵,例如134L 水解酶对应着130 × 130 的加权邻接矩阵,而107M 氧转运蛋白对应着154 × 154 的加权邻接矩阵。同时,不同阶数加权邻接矩阵无法确定样本的中心,从而无法计算聚类簇之间的中心距离,但是可以将2 个聚类簇中样本间的平均距离、最大距离或最小距离作为聚类簇之间的距离。因此,本实验采取层次聚类算法对选取的60个蛋白质样本进行功能聚类。
根据定义1 计算60 个蛋白质样本对应的距离矩阵,并绘制该距离矩阵的热图,如图2A 所示。从图2A 可以看出:通过适当调整顺序,样本大致分为3 个簇,即名为XXXM 的氧转运蛋白、名为2ZXX 的氧转运蛋白和名为XXXL 的水解酶。然后,通过两聚类簇中样本间的平均距离作为聚类簇之间的距离对样本集进行层次聚类,并绘制聚类树,如图2B所示。从图2B 可以看出:选择距离为2.006 作为阈值,产生4 个聚类簇。表1 展示了4 个聚类簇的聚类结果,与图2A 所示结果保持一致。

表1 聚类树产生的4个聚类簇情况表
以两聚类簇中样本间的最大距离作为聚类簇之间的距离,可将样本集聚成如图3A所示的2个簇,其中一个簇由表1中第Ⅰ簇和第Ⅱ簇聚合而成;另一个簇由表1中第Ⅲ簇和第Ⅳ簇聚合而成。将2个聚类簇的结果与真实标签进行比较,并计算其Jaccard 系数、FM指标和Rand指标,如表2所示。聚类理论表明:其Jaccard系数、FM指标和Rand指标越接近1,聚类结果越理想。综合3个指标,本次聚类结果并不很理想。
由于邻接矩阵包含较少信息,无法将表1 中第Ⅰ簇和第Ⅳ簇聚合成一个新簇以代表水解酶,也无法将表1中第Ⅱ簇和第Ⅲ簇聚合成另一个新簇以代表氧转运蛋白。进一步,取样本加权图对应的加权拉普拉斯矩阵作为其矩阵表示,并通过两聚类簇中样本间的平均距离作为聚类簇之间的距离进行层次聚类,将样本集聚类为2 个簇。图3B 展示了其聚类结果,表2 也给出了其对应的聚类指标,其Jaccard 系数、FM 指标和Rand 指标均达到1。聚类结果表明:基于样本加权图对应的加权拉普拉斯矩阵,以两聚类簇中样本间的平均距离作为聚类簇之间的距离进行层次聚类能得到理想的聚类结果。
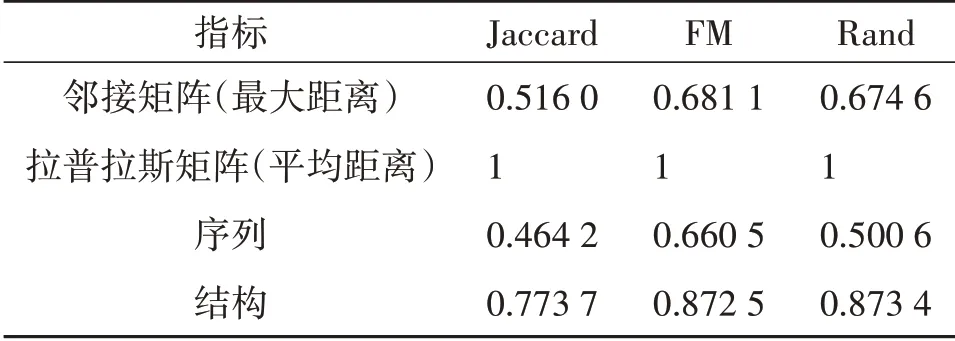
表2 层次聚类各指标信息表

图2 样本距离矩阵热图及基于邻接矩阵以样本间平均距离聚类对应的聚类树

图3 基于邻接矩阵(拉普拉斯矩阵)以样本间最大距离(平均距离)聚类对应的聚类树
最后,将本文提出的相似性度量与序列相似性度量、结构相似性度量进行对比。通过于祥田[20]提供的描述符获得56 维特征向量,并用欧几里得距离度量这60 个蛋白质样本的序列相似度。同时,用RMSD 方法度量这60 个蛋白质样本的结构相似度。图4 展示了其聚类结果,表2 也给出了其对应的聚类指标。对比发现:本文基于样本加权图对应的加权拉普拉斯矩阵所定义的不同维度样本相似性度量是目前最有效的相似性度量方法。

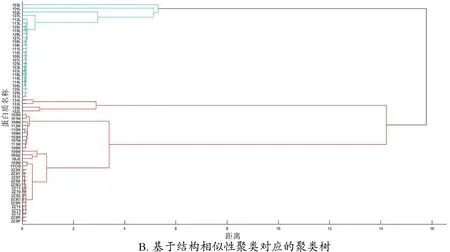
图4 基于序列相似性和结构相似性聚类对应的聚类树
4 结论
本文基于Cheng-范数,提出一种不同维度样本相似性度量新方法。同时,以蛋白质功能聚类为例,通过该方法分别基于蛋白质加权图对应的加权邻接矩阵和加权拉普拉斯矩阵对蛋白质样本进行相似性度量,进一步采用层次聚类算法对其进行聚类。通过综合分析3个聚类指标及聚类树,可以得出:基于样本加权图对应的加权拉普拉斯矩阵,以两聚类簇中样本间的平均距离作为聚类簇之间的距离进行层次聚类能够按照蛋白质功能给出理想的聚类结果,充分表明了该相似性度量方法切实有效。
本文对蛋白质样本进行功能聚类时仅采用了样本加权图对应的加权邻接矩阵和加权拉普拉斯矩阵作为其矩阵表示。实际上,样本加权图对应的点特征矩阵和边特征矩阵包含更多样本结构信息。如果对多种功能的样本进行功能聚类时,采用样本加权图对应的点特征矩阵和边特征矩阵作为其矩阵表示,可能取得更理想的聚类结果。由于本文采用样本加权图对应的加权拉普拉斯矩阵作为其矩阵表示,已经取得理想的聚类结果,所以不再考虑将其他矩阵作为其矩阵表示。
猜你喜欢
北京航空航天大学学报(2022年8期)2022-08-31
上海文化(文化研究)(2022年3期)2022-06-28
南京理工大学学报(2022年1期)2022-03-17
汽车实用技术(2022年4期)2022-03-07
现代英语(2021年18期)2021-11-22
华东师范大学学报(自然科学版)(2019年5期)2019-11-11
作文与考试·初中版(2019年15期)2019-04-28
江西教育B(2019年2期)2019-04-12
华东师范大学学报(自然科学版)(2018年3期)2018-05-14
雪莲(2017年2期)2017-05-12
