
基于QIIME2平台扩增子测序分析探究肉牛瘤胃和直肠中厌氧真菌区系结构的差异
2022-09-01 02:12张亚伟陶薪燕张月娇王月红张元庆
动物营养学报 2022年8期
张亚伟 陶薪燕 张月娇 王月红 张元庆
(山西农业大学动物科学学院,晋中 030801)
新毛孢菌门(Neocallimastigomycota)真菌广泛存在于草食动物尤其是大型哺乳动物的肠道微生态系统中,是一类严格厌氧型真菌。它们凭借自身强大的纤维降解酶体系[1],可以分别提高7%~9%的肠道纤维消化率和高达40%的饲粮采食量[2],因此在反刍动物营养研究中受到普遍关注,近年来也在生物技术领域得到了广泛的研究[3]。截至目前,共有20个属的厌氧真菌得以鉴定和命名[4-6],而非培养学基础的环境分子微生物学研究表明,尚存在更多属水平的厌氧真菌有待分离、鉴定和描述[7]。随着研究的不断深入,许多研究发现,肠道厌氧真菌在分类学或功能水平上的变化与甲烷生成或剩余采食量密切相关[8-9],因此准确评估肠道厌氧真菌的区系组成和多样性对评价其与宿主动物生产性能之间的联系至关重要。
迄今,最常用的探究肠道乃至环境中微生物区系组成和多样性的方法是标记基因基础的扩增子测序,即宏分类组学分析[10]。内转录间隔区1(ITS1)是环境中厌氧真菌分类学和多样性研究常用的标记基因区域,然而厌氧真菌中该基因区长度的同源异质性和单核苷酸多态性的存在,可能导致经典的以操作分类单元(operational taxonomic unit,OTU)为基础的分类学分析方法产生很大的误差,同时也会受其他高通量数据处理过程的影响[6,11]。QIIME2是一个免费、开源且可扩展的微生物组信息学分析平台,它可以完成从原始测序数据至统计分析的全部微生物组高通量数据处理过程,同时可以实现单体代表性序列基础的分类学注释和分析[12],后者在一定程度上可以减少序列相似性聚类产生OTU过程导致的误差。然而,目前尚没有针对肠道厌氧真菌群落的QIIME2平台扩增子测序分析流程的研究报道。
另外,许多研究表明,碳源和宿主品种对厌氧真菌的区系结构具有显著的影响,即厌氧真菌群落产生了生态位分化[13-14]。也就是说,在不同的生态位中存在着不同的优势厌氧真菌,具有不同的生物学特性,同时发挥着不同的生态学功能和作用。然而,迄今我们对其生物学特性的理解多来自于瘤胃厌氧真菌,因此了解厌氧真菌在草食动物前后肠道内区系结构的差异,将有助于加深对厌氧真菌生物学特性及其在肠道微生态营养中作用的理解,例如不同种类的厌氧真菌菌体或孢子(囊)被宿主动物消化的程度是否相同等。为此,本研究拟介绍一种基于QIIME2平台的厌氧真菌扩增子测序分析流程,并探究肉牛瘤胃和直肠中厌氧真菌区系结构的差异。
1 材料与方法
1.1 试验动物管理和样品采集
选择6头14月龄、装有永久性瘤胃瘘管的晋南牛[体重(350±20)kg]作为试验牛,饲养于山西农业大学动物科学学院畜牧兽医研究所试验基地。所有试验用瘘管牛均饲喂相同的饲粮,该饲粮由75%的全株玉米青贮和25%的精料补充料混合配制而成(干物质基础)。精料补充料购自山西易大饲料有限公司,由玉米、菜籽粕、糖蜜、石粉、磷酸氢钙、氯化胆碱、蛋氨酸、复合维生素和复合微量元素等原料配制而成。全混合日粮的主要营养成分含量为粗蛋白质10.9%、粗脂肪3.1%、粗纤维24.9%、无氮浸出物52.3%和粗灰分8.7%(干物质基础)。试验动物每日08:00和17:00各饲喂1次,自由饮水。试验动物的饲养管理及后续的样品采集方案均得到了山西农业大学实验动物伦理委员会的批准。
预饲15 d后,于第16天晨饲前分别经瘤胃瘘管和直肠直接采集6头试验牛的肠道内容物样品,所采集的瘤胃内容物和直肠粪便均为原始样品,不进行固液分离。所有样品均直接置于已灭菌离心管中,随即液氮冷冻,-80 ℃保存备用。
1.2 DNA的提取
瘤胃内容物和直肠粪便样品中全基因组DNA的抽提参照Yu等[15]的方法,使用粪便微生物DNA提取试剂盒(Omega Bio-Tek,Norcross,GA,美国)进行。DNA的纯度和浓度使用NanoDrop2000(NanoDrop Technologies,Wilmington,DE,美国)进行检测,完整性使用10 g/L琼脂糖凝胶电泳进行分析。纯度和完整性合格的DNA样品方可用于后续扩增子文库构建和高通量测序。
1.3 扩增子文库构建和高通量测序
为了特异性地评估肠道厌氧真菌的群落结构,本研究选用厌氧真菌特异性引物对[16-17]MN100F(5′-TCCTACCCTTTGTGAATTTG-3′)和MNGM2R(5′-CTGCGTTCTTCATCGTTGCG-3′)对真菌ITS1区域进行PCR扩增。扩增过程在PCR循环温控仪(ABI GeneAmp®9700,Applied Biosystems,新加坡)中进行,具体程序如下:94 ℃预变性2 min,然后进行35个温控循环(94 ℃变性30 s,52 ℃退火30 s,72 ℃延伸30 s),最后72 ℃稳定延伸7 min。PCR反应体系为:2×Pro Taq预混液10 μL,上、下游引物溶液(5 μmol/L)各0.8 μL,模板DNA 10 ng,最后用ddH2O补足至20 μL。每个样本3个重复。
将同一样本的3个PCR重复产物混合并使用20 g/L琼脂糖凝胶电泳进行富集,随后切取目的条带使用DNA凝胶提取试剂盒(Axygen Biosciences,Union City,CA,美国)进行回收纯化。纯化后的扩增子样品浓度使用荧光定量仪(QuantusTM,Promega Corporation,美国)进行检测,大小和完整性使用20 g/L琼脂糖凝胶电泳进行检测。条带大小正确、浓度合适的扩增子样品方可进行后续的建库和测序操作。
扩增子测序文库使用NEXTflexTM快速DNA测序试剂盒(Bioo Scientific,美国)按照厂家使用说明进行构建,构建好的测序文库使用Illumina MiSeq PE300平台(Illumina Inc.,San Diego,CA,美国)进行高通量测序,产生长度为300 bp的双端序列文件用于后续分析。测序过程由上海美吉生物医药科技有限公司完成。
1.4 测序数据处理
扩增子测序数据使用QIIME2平台(版本2021.08)进行处理,具体流程参见图1。首先,使用q2-tools插件中的import方法将下机数据集导入至QIIME2平台中;随后,使用q2-cutadapt插件中的trim-paired方法在默认条件下将barcode和primer碱基序列从双端序列中移除[18];最后,使用q2-dada2插件中集成的denoise-paired方法对双端序列进行降噪、合并、去嵌合体和去重复等处理[19],该过程设置参数为移除双端序列中第203位后的所有碱基序列(由于测序质量下降),并获得代表性序列(amplicon sequence variants,ASVs)及其频率分布表。
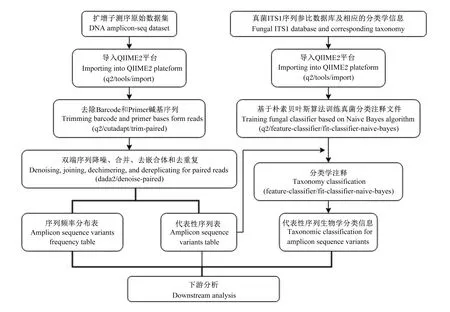
图1 厌氧真菌扩增子测序数据处理流程
本研究采用机器学习的方法进行ASVs的分类学注释[20],参比数据库及分类学注释信息选用UNITE真菌ITS数据库[21](版本8.3,2021-10-05发布)。具体过程如下:首先使用q2-tools插件中的import方法将99% OTU参比数据集和相应的分类学注释信息文档导入至QIIME2平台,随后使用q2-feature-classifier插件中集成的fit-classifier-naive-bayes方法将二者训练成朴素贝叶斯分类注释文件[22]。然后,基于该注释文件,根据机器学习的原理,使用q2-feature-classifier插件中的classify-sklearn方法对ASVs进行分类学注释[22]。ASVs及其频率分布和分类学注释信息用于下游多样性和统计分析。
1.5 菌群多样性分析
对于菌群多样性分析,首先基于1.4得到的ASVs利用q2-phylogeny插件中集成的align-to-tree-mafft-fasttree处理流程生成系统发育树[23];随后利用q2-diversity插件中集成的alpha-rarefaction方法分析alpha多样性指标随测序深度变化的趋势(即稀疏度曲线分析),并设置最大测序深度为33 627,以确保所有样品的测序深度足以用于下游分析菌群多样性。随后,以系统发育树和1.4得到的ASVs频率分布表为基础,利用q2-diversity插件中集成的core-metric-phylogenetic方法流程计算香农指数(Shannon index)、费氏系统发育学多样性指数(Faith PD)、可观测特征序列(observed futures)、辛普森指数(Simpson index)和皮氏均匀度指数(Pielou evenness)等alpha多样性指标和Unweighted UniFrac和Weighted UniFrac距离等beta多样性指标,该过程设置所有样品的测序深度均为33 627。
1.6 统计分析
厌氧真菌生物学分类信息在属水平上进行分析,并定义相对丰度在至少1个样本中大于0.01%的菌属是可被鉴定的,而平均相对丰度大于0.1%且在每个组半数以上的样本中存在的菌属是可被检测的,只有可被检测的菌属才被用于组间差异分析。菌群alpha多样性参数和属水平相对丰度的组间差异采用Kruskal-Wallis检验进行分析,显著性水平为P<0.05,当0.05≤P<0.10时认为有显著性变化趋势,该过程使用R(版本4.1.2)中的rcompanion软件包(版本2.4.13)完成。此外,各样本beta多样性的组间差异使用主坐标分析(PCoA)进行分析和展示,显著性采用置换多元方差分析(PERMANOVA)进行检验[24],该过程使用qiime2R软件包(版本0.99.20)完成。
2 结果与分析
2.1 测序情况介绍
扩增子测序共产生813 343对双端序列,平均每个样本(67 779±10 392)对双端序列。经序列降噪、合并和去嵌合体后,共产生676 759条、平均(56 397±9 867)条有效ITS1序列,用于后续分析。有效序列经去除重复序列后,共产生256条代表性ITS1序列,即ASVs,序列平均长度(233±16)个碱基。所有样本的覆盖度指数(Good’s coverage)均为1.000,表明本研究的测序深度足以覆盖全部厌氧真菌群落。该结果也可由alpha多样性稀疏度曲线分析(图2)进一步证明:当测序深度大于15 000条序列时,所有样本的香农指数和费氏系统发育学多样性指数的稀疏度曲线均趋于水平,即厌氧真菌物种丰度和系统发育学多样性均不再增加,而该值远小于单样本最小有效ITS1序列数(33 627),表明所有样本的测序深度均足以覆盖全部厌氧真菌群落。
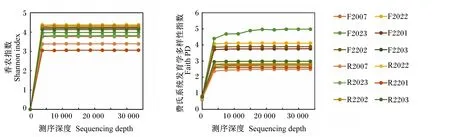
F2007、F2022、F2023、F2201、F2202和F2203代表直肠粪便样本,R2007、R2022、R2023、R2201、R2202和R2203代表瘤胃内容物样本。
2.2 肠道内容物采样位点对厌氧真菌群落多样性的影响
表1列出了瘤胃和直肠内容物中厌氧真菌群落的alpha多样性指标。由表可知,除了费氏系统发育学多样性指数外,其他厌氧真菌群落alpha多样性指标在瘤胃和直肠之间均没有统计学显著差异(P>0.05);直肠粪便中厌氧真菌群落费氏系统发育学多样性指数显著大于瘤胃内容物(P<0.05)。
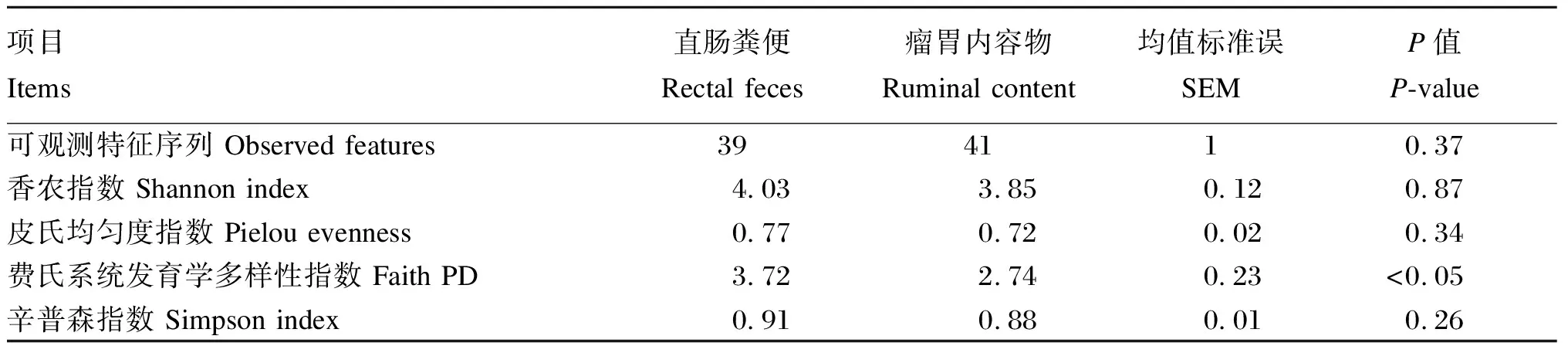
表1 不同肠道位点厌氧真菌群落的alpha多样性指标
主坐标分析显示,基于Unweighted UniFrac(图3-A)和Weighted UniFrac(图3-B)距离的主坐标1(PCo1)和主坐标2(PCo2)共分别能解释总变异的70.20%和69.92%;同时,基于2种距离得到的瘤胃内容物和直肠粪便样品真菌区系结构明显分离,该现象也由PERMANOVA分析结果(P<0.05)进一步证实。值得注意的是,瘤胃内容物中厌氧真菌群落beta多样性置信椭圆均小于直肠粪便样品,即厌氧真菌群落结构在瘤胃中的变异程度更低。
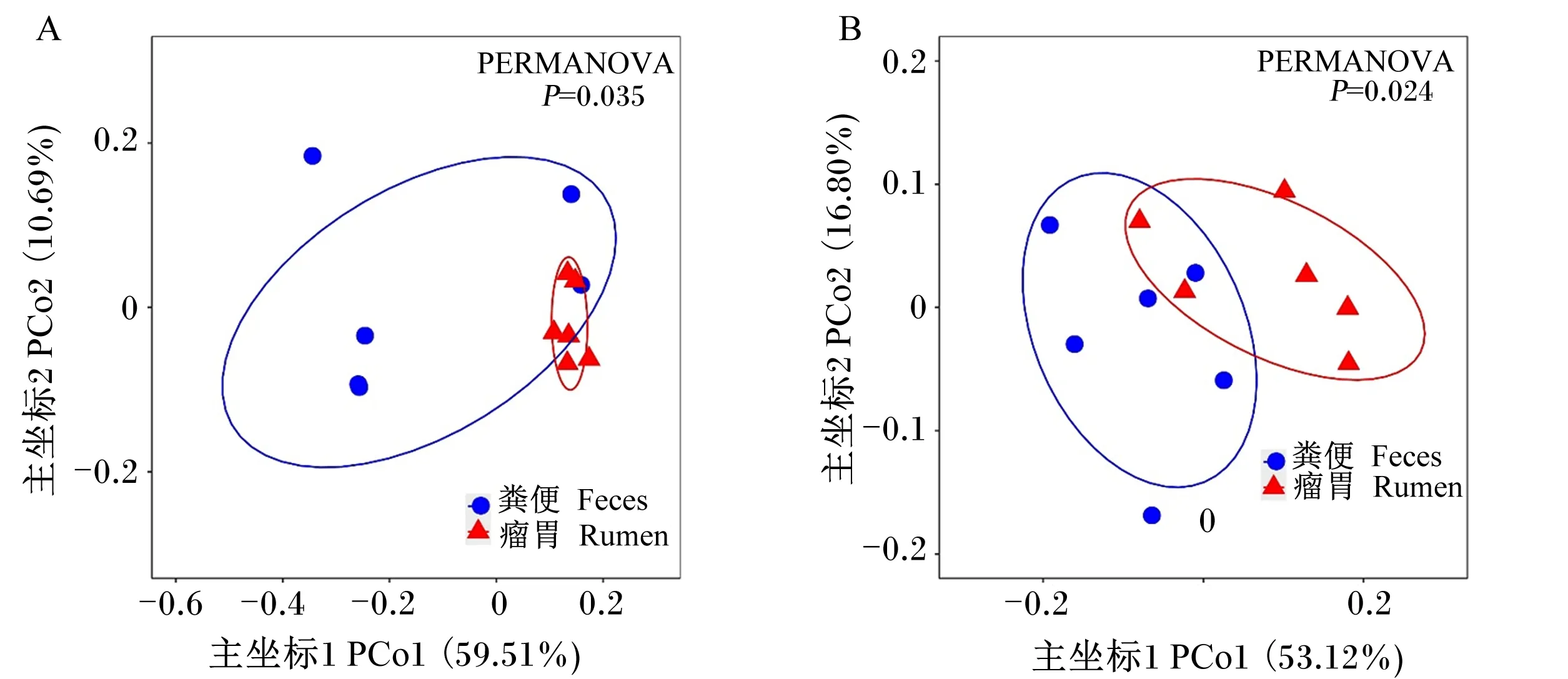
PERMANOVA即置换多元方差分析;蓝色和红色椭圆分别代表粪便和瘤胃内容物厌氧真菌群落beta多样性的95%置信区间。
2.3 肠道内容物采样位点对属水平厌氧真菌相对丰度的影响
菌群注释分析显示,本试验共从肉牛肠道内容物样品中鉴定出7个属水平肠道真菌群落,均处于可检测水平。其中,厌氧真菌属(Anaeromyces)、瘤胃壶菌属(Piromyces)、盲肠真菌属(Caecomyces)、肠道真菌属(Cyllamyces)、奥宾氏真菌属(Orpinomyces)和未归类新毛孢菌科(unclassified Neocallimastigaceae)等6个属水平菌群为已确定厌氧真菌,分别占真菌群落总量的23.3%、16.8%、27.6%、25.6%、5.4%和0.6%。另外,盲肠真菌属、肠道真菌属、厌氧真菌属和瘤胃壶菌属为优势菌属,共约占全部厌氧真菌群落的93.2%。差异丰度分析显示,直肠粪便样品中盲肠真菌属和未归类真菌(unclassified fungi)的平均相对丰度分别为33.7%和1.7%,较瘤胃内容物样品中的21.4%和0具有升高的趋势(0.05≤P<0.10);更为严格的线性判别效应尺寸分析(LEfSe)显示,上述2个菌群的线性判别分析(LDA)得分均大于4.0,但仅未归类真菌的相对丰度差异达到了显著性水平(P<0.05)。
3 讨 论
本研究介绍了一种特异性的分析肠道厌氧真菌区系结构的扩增子分析流程,该流程基于QIIME2平台,利用DADA2对高通量数据进行降噪以获得所有单体代表性序列集,并利用该代表性序列集基于机器学习算法进行分类学注释。与经典的OTU聚类模式分析流程相比,本研究所用流程既不需要独立进行序列过滤、双端序列合并、去嵌合体和去重复序列等数据处理过程,也无需先对单体代表性序列进行聚类以获得OTU后再进行分类学注释,操作相对较为简便,因此由所选数据处理方法不同导致的误差也相对较少,有利于不同研究间的横向对比。更为重要的是,OTU是基于序列相似度进行聚类而产生的,并非生物学意义上的分类水平,在聚类的过程中不可避免的会导致某些原本非同一物种的单体代表性序列被人为的归为同一分类簇,造成系统发育信息丢失[19]。因此,对于同源异质性和单核苷酸多态性普遍存在的厌氧真菌ITS1区域[6,16],OTU聚类过程造成的系统发育分类信息损失理论上会更为严重。本研究所用数据处理流程是在单体代表性序列的基础上进行分类学注释分析的,因此可以在一定程度上减少由于OTU聚类导致的系统发育学分类信息的损失,理论上更适用于肠道厌氧真菌群落结构的分析。值得一提的是,目前有关肠道真菌的研究多使用通用引物将包括需氧真菌在内的所有真菌生物标记物进行全克隆测序[25-27],仅有少数研究专注于厌氧真菌群落[28-29],考虑到肠道内环境特点以及厌氧真菌和需氧真菌各自的生活习性和生物学特性的较大差异,我们认为特异性地构建厌氧真菌测序文库对探究厌氧真菌在草食动物肠道营养中的生物学功能更具意义。
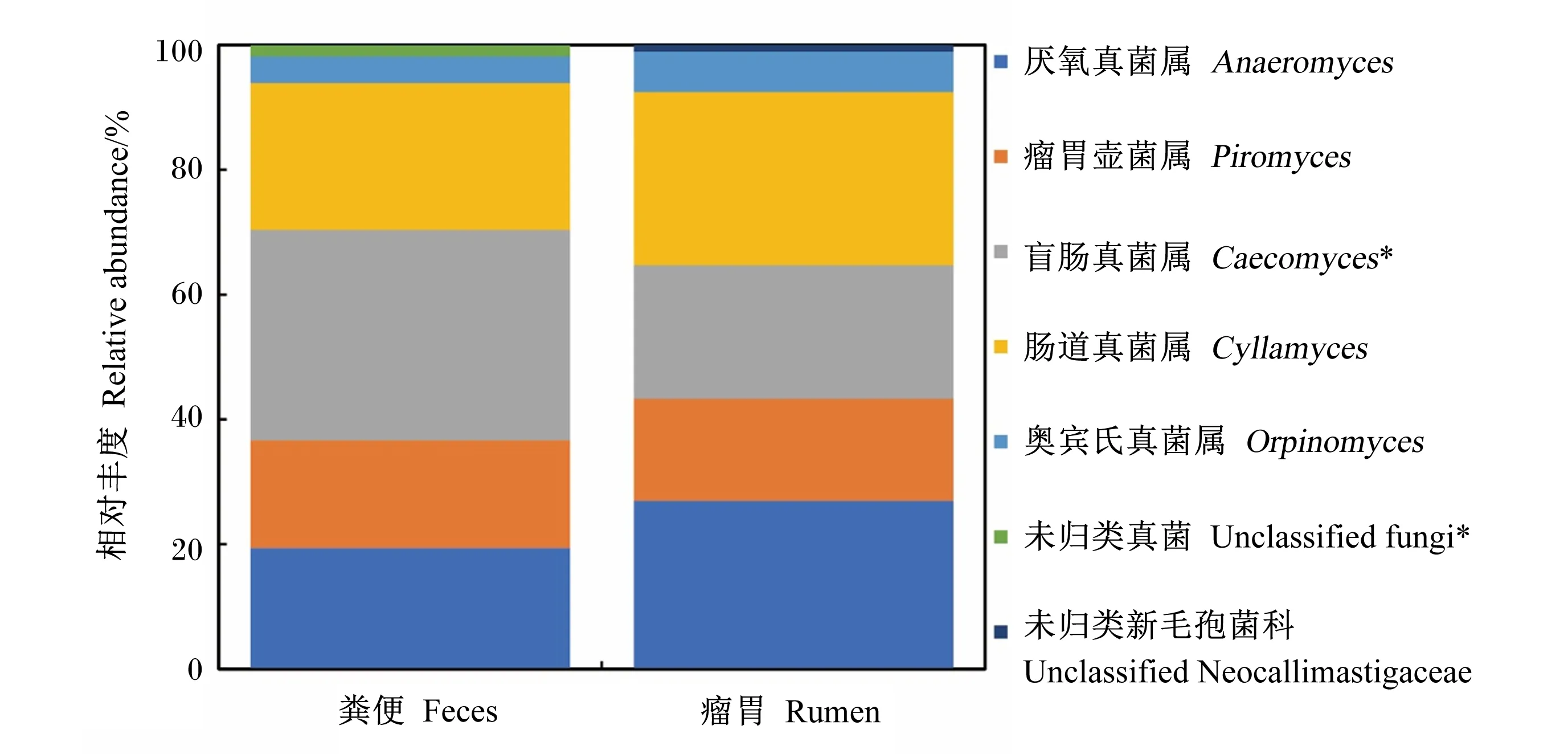
*代表该菌属在瘤胃和直肠内容物中的相对丰度具有差异变化的趋势(0.05≤P<0.10)。
在该分析流程的基础上,本研究利用扩增子测序进一步比较了晋南牛瘤胃和直肠中厌氧真菌区系结构的差异。alpha多样性的组间差异分析表明,厌氧真菌代表性序列的丰富度和均匀度在瘤胃和直肠中均没有统计学差异,说明绝大多数的瘤胃真菌均可能存在特殊的休眠结构[30],该结构可以抵抗肠道消化过程而到达后肠道并再次萌发和定植。同时,直肠中真菌群落的系统发育学多样性要显著高于瘤胃中,即直肠中真菌菌群间的亲缘关系相对较远;此外,beta多样性的组间差异分析证实,瘤胃和直肠内容物中真菌区系结构具有统计学差异。鉴于瘤胃和直肠内环境的差异,本研究结果进一步说明,除了可利用碳源[13]和宿主动物类型[14,29]外,即便是同一动物个体的前后肠道中,真菌群落也表现出了明显的生态位分化。与本研究结果相似,Mura等[31]发现,厌氧真菌群落结构在马消化道的不同区域间也具有明显的差异。类似的生态位分化现象在细菌等其他类型肠道微生物中也有报道[32]。
由菌群注释分析得到的属水平厌氧真菌中,瘤胃壶菌属和盲肠真菌属为单中心厌氧真菌,共分别占瘤胃和直肠内容物真菌群落的37.8%和50.9%;厌氧真菌属、肠道真菌属和奥宾氏真菌属为多中心厌氧真菌,共分别占瘤胃和直肠内容物真菌群落的61.2%和47.3%,表明多中心型厌氧真菌在瘤胃内环境中数量相对更多,而单中心型厌氧真菌则更适应于后肠道内环境。结合直肠中盲肠真菌属的相对丰度较瘤胃中有升高的趋势,同时考虑到反刍动物后肠道内容物中较高比例的难降解饲料组分,提示单中心厌氧真菌尤其是盲肠真菌属可能有用作肉牛直饲型微生物或厌氧发酵饲料(如青贮饲料)接种剂进而改善饲料消化的潜力。与本研究结果相似,Guo等[29]在牦牛瘤胃中也鉴定出了本研究中所有注释出的属水平厌氧真菌,同时还鉴定出了新毛孢菌属(Neocallimastix)和骆驼真菌属(Oontomyces)的存在;彭全辉等[27]在西杂牛和犏牛杂交牛的瘤胃中也鉴定到了瘤胃壶菌属、奥宾氏真菌属和新毛孢菌属的存在。然而与本研究不同的是,这2项研究中优势菌属分别为瘤胃壶菌属和新毛孢菌属,产生这种差异的原因可能是测序文库构建和数据处理方法的差异,以及宿主动物类型和所采食饲粮的不同所致。
4 结 论
① 本研究介绍了一种适用于肠道厌氧真菌扩增子测序数据的处理流程,该流程简便且可操作性强,便于研究结果间的横向比较,同时理论上可以减少经典的OTU聚类方法导致的系统发育学信息丢失。
② 厌氧真菌群落在肉牛瘤胃和直肠中具有一定的生态位分化,该现象主要是由盲肠真菌属为主的单中心厌氧真菌所引起的。
猜你喜欢
当代水产(2022年8期)2022-09-20
中国音乐学(2022年2期)2022-08-10
基层中医药(2022年1期)2022-07-22
中国饲料(2022年5期)2022-04-26
昆明医科大学学报(2022年2期)2022-03-29
现代临床医学(2022年1期)2022-02-12
家庭医药·快乐养生(2021年8期)2021-08-30
学校教育研究(2020年7期)2020-04-09
农民致富之友(2019年25期)2019-09-03
科学种养(2009年6期)2009-06-03
