
马铃薯卷叶病毒与S病毒重组双CP多克隆抗体的制备
2022-09-14 04:58宋志成韦晓玲隋炯明郭宝太
华北农学报 2022年4期
吉 祥,宋志成,韦晓玲,杨 煜,隋炯明,郭宝太
(1.青岛农业大学 农学院,山东省旱作农业技术重点实验室,山东 青岛 266109;2.山东省农业科学院 蔬菜花卉研究所,山东 济南 250100)
马铃薯是重要的粮菜兼用的作物,病毒侵染可导致马铃薯产量与品质的明显下降,病毒病危害的防治是马铃薯生产的基本要求。通过茎尖培养结合严格的病毒检测可获得高质量脱毒种薯,而脱毒种薯的利用是防治马铃薯病毒危害的有效手段。茎尖培养脱毒的具体方法有多种,病毒检测的主要方法是血清学方法与分子生物学方法[1]。
马铃薯卷叶病毒(Potatoleafrollvirus,PLRV)与S病毒(PotatovirusS,PVS)是2种危害马铃薯的主要病毒。PLRV是马铃薯卷叶病毒属(Polerovirus)的代表成员,病毒粒体为球状等轴对称结构,其侵染可导致马铃薯严重减产,甚至达到80%,但通过茎尖培养脱除PLRV比较容易。PVS是香石竹潜隐病毒属 (Calavirus)的成员,病毒粒体为线条状,PVS单独侵染造成的产量损失为10%~20%,但与马铃薯X病毒(PVX)混合侵染可造成更严重的危害[2]。PVS是世界马铃薯产区广泛分布的病毒,脱除非常困难,脱毒种薯种植后容易再次感染病毒。脱毒种薯生产、利用中针对PLRV与PVS的检测都非常必要。
酶联免疫吸附测定(Enzyme-linked immuosorbent assay,ELISA)是植物病毒检测的基本方法,1977年Casper首先将该法用于马铃薯病毒检测,双抗体夹心(DAS)ELISA具有操作简单、特异性强及灵敏度高的特点,可以有效地用于马铃薯脱毒种薯生产与利用中的病毒检测[3]。
利用马铃薯病毒外壳蛋白(Coat protein,CP)基因原核表达的重组CP作抗原可以制备马铃薯病毒特异性抗血清(抗体),陆续有成功的报道。由于PLRV-CP基因原核表达困难,Plchova等[4]采用有利于精氨酸稀有密码子表达的特殊受体菌才实现了该基因的有效表达并将重组CP抗血清(抗体)用于PLRV的间接ELISA检测。王韬远等[5]通过改造PLRV-CP基因的密码子,实现了该基因的原核表达并利用重组CP制备出了高效价PLRV特异性抗血清(抗体)。青岛农业大学遗传研究室采用删除PLRV-CP基因中抑制表达序列的策略实现了该基因的高效原核表达[6],并将制备的重组CP多克隆抗体与酶标抗体用于PLRV的DAS-ELISA检测[7]。2004年李广存等[8]克隆了PVS-CP基因,利用重组CP制备出了PVS特异性抗血清(抗体)并应用于ELISA检测,但针对PVS只见到这一例研究结果。利用重组CP制备马铃薯病毒特异性抗血清(抗体)将为我国包括PLRV与PVS在内的马铃薯病毒抗血清(抗体的)大量制备及ELISA试剂盒的国产化提供有力的技术支撑。用一种重组CP制备的抗体只能检测一种病毒,这是其局限性。
本研究的目的是通过原核表达PLRV-CP基因与PVS-CP基因序列形成的融合基因,获得高纯度PLRV与PVS重组双CP,并将重组双CP多克隆抗体用于PLRV与PVS这2种病毒的间接ELISA检测与DAS-ELISA检测,旨在为提高脱毒种薯病毒ELISA检测效率、降低检测成本奠定基础。
1 材料和方法
本研究于2016—2020年在青岛农业大学遗传研究室完成。
1.1 质粒与菌种
马铃薯卷叶病毒CP基因原核表达载体pET22b-LRCP及PVS-CP基因克隆载体pMD18-SCP由本研究室构建,大肠杆菌DH5α与BL21菌株由本研究室保存。
1.2 主要试剂与试剂盒
限制性内切酶Hind Ⅲ、XhoⅠ、T4DNA连接酶及DNA Marker购自大连宝生物公司。质粒DNA小量提取及DNA胶回收试剂盒、氨苄青霉素、IPTG、细菌总蛋白提取试剂、SDS-PAGE试剂、蛋白Marker、羊抗兔HRP酶标二抗、ELISA酶标板、透析膜等购自上海生工公司。金属离子亲和层析柱(HiTrap Chelating HP)、抗体纯化柱(HiTrap Protein G HP)购自GE Healthcare公司。马铃薯Y病毒(PVY)、A病毒(PVA)、M病毒(PVM)及PLRV、PVS、PVX等6种主要病毒的阳性标准物与阴性标准物购自美国Agdia公司。
1.3 PLRV与PVS融合双CP基因原核表达载体的构建
用Hind Ⅲ与XhoⅠ酶切质粒pMD18-SCP,回收含有PVS-CP基因序列的DNA片段,并将该片段定向插入到pET22b-LRCP中PLRV-CP基因下游,构建出了PLRV与PVS融合双CP基因的原核表达载体,经酶切鉴定与DNA测序确认表达载体的正确性,该表达载体命名为pET22b-LRCP/SCP。
1.4 融合双CP基因原核表达的诱导
将重组菌BL21(pET22b-LRCP/SCP)单菌落接种于LB液体培养基进行活化培养,取200 μL活化的菌液接种于20 mL LB液体培养基中,37 ℃振荡培养,至菌液OD600值达到0.5~0.8。固体与液体LB培养基中氨苄青霉素含量均为100 μg/mL。取出1 mL菌液加入EP管中继续培养作为未经诱导的对照,向剩余的菌液中加入IPTG至终浓度为1 mmol/L,37 ℃振荡培养8 h后,SDS-PAGE电泳检测诱导表达的结果。
1.5 菌体蛋白的提取与重组双CP的纯化
菌体蛋白的提取采用了2种方法:溶菌酶法与超声破碎法。溶菌酶法中,用裂解液(150 mmol/L Tris-HCl,1.5 mol/L NaCl)悬浮菌体,用溶菌酶裂解菌体,加PMSF至终浓度为1 mmol/L抑制蛋白酶活性,加DNase Ⅰ降解细菌DNA,最后获得包涵体,具体方法见隋炯明等[6]的描述。超声破碎法中,每50 mL菌液离心获得的菌体用4 mL预冷的0.1 mmol/L的PBS缓冲液悬浮,冰上用超声波细胞粉碎机(新芝JY92-ⅡN)超声破碎18个循环后,放置3 min,再继续超声破碎。超声破碎参数:100%振幅,3 s运行,6 s停止,35个循环。4 ℃ 10 000 r/min 离心45 min获得包涵体蛋白。
2种方法获得的包涵体都用含8 mol/L尿素的结合缓冲液(20 mmol/L NaH2PO4,0.5 mol/L NaCl,8 mol/L尿素,30 mmol/L咪唑,pH值7.5)溶解。溶解液经0.22 μm滤膜过滤后采用1 mL的镍离子亲和层析柱纯化,上样与洗涤后利用洗脱液(20 mmol/L NaH2PO4,0.5 mol/L NaCl,8 mol/L尿素,500 mmol/L咪唑,pH值7.5)洗脱并收集目的蛋白。
重组双CP蛋白诱导表达、提取与纯化的结果通过SDS-PAGE检测确认。蛋白样品的制备方法是:取未经诱导与诱导过的菌液各1 mL,12 000 r/min离心2 min,弃上清,菌体沉淀用40 μL 1×上样缓冲液悬浮;分别取包涵体悬浮液、目的蛋白纯化溶液20 μL,加入5 μL 5×上样缓冲液并混匀。上述混合液均煮沸处理10 min,冷却后4 ℃静置5 min,取10 μL上清点样。经洗脱获得的高纯度重组双CP溶液先用不同浓度尿素的PBS透析,再用PEG 6000粉末处理浓缩,最后采用Bradford法进行定量。
1.6 抗血清的制备及抗体的分离与纯化
用高纯度重组双CP作抗原免疫新西兰大白兔制备抗血清,效价测定采用间接ELISA法,抗血清制备由上海生工公司完成。采用饱和硫酸铵沉淀法从抗血清中分离多克隆抗体(IgG),抗体粗分离物用1 mL Protein G亲和层析柱纯化。用10 mL磷酸盐缓冲液(20 mmol/L Na3PO4,pH值7.0)洗涤,最后用1.5 mL洗脱液(0.1 mol/L Glycine-HCl,pH值2.7)洗脱收集抗体,并用缓冲液(1 mol/L Tris-HCl,pH值8.8)中和,中和后的抗体可直接用于间接ELISA测定,经过透析后才可以进行酶标记。
1.7 重组双CP抗体活性与特异性的测定
重组双CP多克隆抗体与PLRV、PVS反应活性的测定采用间接ELISA法。具体步骤是:用抗原提取缓冲液(0.01 mmol/L Na2SO3,2% PVP-4000,0.2% BSA,2% Tween-20的PBS,pH值7.4)溶解病毒阳性标准物制成抗原包被工作液,以病毒阴性标准物包被反应孔作为阴性对照,以抗原提取缓冲液为空白对照,每反应孔加100 μL包被酶标板,37 ℃培育2 h。甩去抗原包被工作液,用PBST洗板,每孔加200 μL,静置3 min后甩去,洗板3次。在滤纸上甩干反应孔,用3% BSA封闭,每孔加200 μL,37 ℃孵育1.5 h。甩去封闭液,洗板方法同上。用抗体包被缓冲液稀释纯化的多克隆抗体,每孔加100 μL,37 ℃孵育2 h,甩去抗体稀释液后洗板。用酶标抗体缓冲液按照1∶2 000稀释酶标二抗(羊抗兔IgG-AP)制成酶标抗体工作液,每孔100 μL,37 ℃孵育2 h,甩去酶标抗体工作液后洗板。配制TMB显色工作液,每孔加100 μL,静置3~5 min,最后加入100 μL硫酸终止反应。目测显色结果,并用BioTek公司Elx800型酶标仪测定OD450吸收值,用样品孔OD450≥2.5倍阴性孔OD450作为阳性判断的标准。
同样用上述间接ELISA方法测定重组双CP多克隆抗体与6种马铃薯主要病毒阳性标准物的反应特异性,为了增强反应信号,重组CP多克隆抗体按照1∶500比例稀释,酶标二抗(羊抗兔IgG-AP)按照1∶1 000的比列稀释。
1.8 抗体的酶标记
重组双CP多克隆抗体(IgG)的碱性磷酸酶标记采用戊二醛一步反应法,酶标抗体活性的测定采用直接ELISA法。抗血清制备、抗体的分离与酶标记以及酶标抗体活性测定的具体方法见参考文献[7,9]。
1.9 PLRV与PVS的DAS-ELISA检测
以免疫前抗血清为阴性对照(阴性对照1),以抗原提取缓冲液为空白对照,重组CP(1 μg/mL)作为阳性对照,病毒阴性物作为抗原的阴性对照(阴性对照2)。多克隆抗体按照1∶100、1∶200与1∶300的比例用抗体包液稀释,每反应孔加100 μL,37 ℃孵育2 h。洗板后用BSA进行封闭处理,37 ℃培育2 h。PLRV、PVS阳性标准物用抗原提取液稀释后,每孔加100 μL,37 ℃孵育2 h。最后用酶标抗体缓冲溶液按1∶100的比例稀释酶标抗体(IgG-AP),每个样品孔内加100 μL,37 ℃孵育2 h。抗体包被、封闭、抗原结合以及酶标抗体结合后都进行甩去液体、洗涤与甩干反应孔的处理,方法同1.6。配制pNPP底物显色工作液,每孔加100 μL,室温避光静置10~20 min,然后每孔加入100 μL 的1 mmol/L的NaOH终止反应,观察显色结果,并测定OD405吸收值。
2 结果与分析
2.1 PLRV与PVS重组双CP的原核表达及提取与纯化
将含有PVS-CP基因序列的双酶切片段定向插入pET22b-LRCP,构建出了PLRV与PVS双CP融合基因的原核表达载体,命名为pET22b-LRCP/SCP。Hind Ⅲ单酶切该表达载体的产物是一条DNA带(图1,泳道2),Hind Ⅲ+XhoⅠ双酶切产物是2条DNA带,其中小片段为PVS-CP基因(图1,泳道3—4),单、双酶切片段数与大小都符合预期。测序结果表明,PLRV与PVS双CP融合基因序列正确,大小为1 401 bp,其中PLRV-CP基因的序列(498 bp)在5′端、PVS-CP基因的序列(882 bp)在中间、3′端为载体的部分序列,载体构建符合要求,上述498 bp的序列是缺失突变的PLRV-CP基因[6]。
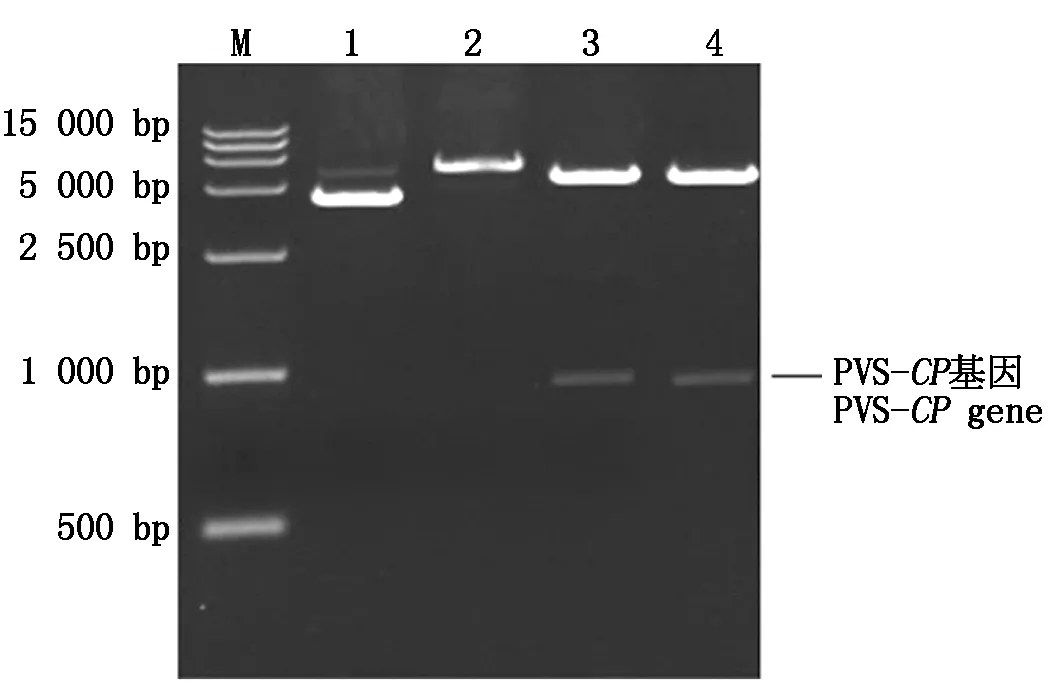
M.DNA Marker 15000;1.表达载体pET22b-LRCP/SCP;2.质粒pET22b-LRCP/SCP+HindⅢ(+);3—4.质粒pET22b-LRCP/SCP+Hind Ⅲ+Xho Ⅰ。
用溶菌酶裂解重组菌BL21(pET22b-LRCP/SCP),菌体蛋白及提取的包涵体蛋白中都有一条分子质量约为51.2 ku的特异性蛋白条带(图2,泳道2—3)。采用试剂盒推荐的结合缓冲液与洗脱缓冲液,用镍离子亲和层析法纯化包涵体蛋白溶解液,洗脱产物中有明显的目的蛋白条带(重组双CP),但也有明显的杂蛋白条带(图2,泳道4)。
改用超声波破碎菌体,提取的包涵体蛋白经过溶解后,溶解液中目的蛋白条带明显,菌体蛋白相对含量很低(图3,泳道3),SDS-PAGE结果显示,纯化产物只有清晰的目的条带(图3,泳道4—5),也就是获得了高纯度的PLRV与PVS的重组双CP。超声波破碎菌体的同时也作用于包涵体,增加了它的可溶性,在结合缓冲液与洗脱液成分及洗涤、洗脱条件不变的情况下,提高了包涵体蛋白的得率与目的蛋白的相对含量,从而降低了目的蛋白纯化难度、获得了高纯度重组双CP。
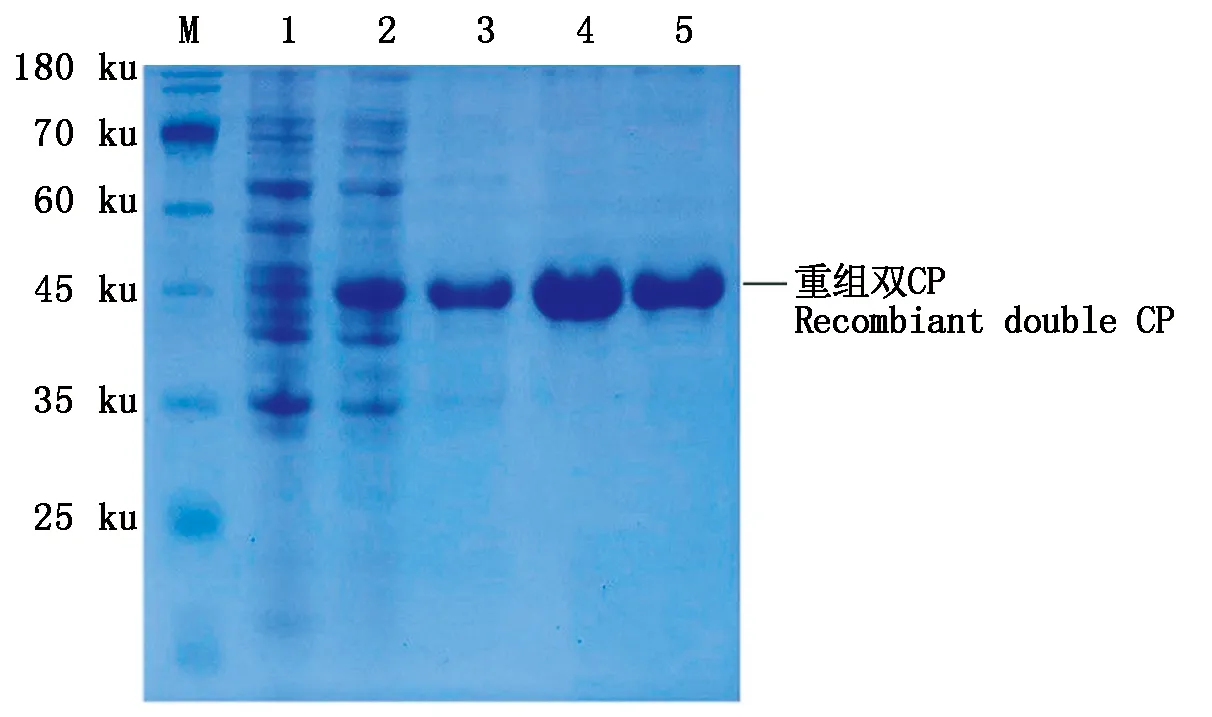
M.蛋白分子量标准;1.未诱导的菌体;2.诱导后的菌体; 3.包涵体蛋白溶解液;4—5.纯化的重组双CP。 M.Protein Marker;1.Uninduced bacterial cell;2.Bacterial cell induced with IPTG;3.Dissolved inclusion body protein; 4—5.Purified recombinant double CP.
2.2 重组双CP抗体的分离、纯化及其活性与特异性的测定
间接ELISA测定表明,获得了PLRV与PVS重组双CP的高效价(1∶128 k)抗血清,抗血清中含有大量杂蛋白(图4,泳道1),饱和硫酸铵沉淀获得的抗体粗分离物中杂蛋白去除非常明显(图4,泳道2),经Protein G纯化获得的抗体的重链与轻链带型清晰,杂蛋白已很少(图4,泳道3),表明已获得了高纯度的重组双CP多克隆抗体(IgG)。
用PLRV阳性标准物包被反应孔、病毒阴性物作为阴性对照,间接ELISA测定显示,阴性及空白对照不显色,抗原包被孔显色,且随着抗体稀释度的增加颜色变浅,结果表明,重组双CP多克隆抗体与PLRV呈特异性反应。根据阳性标准判断,纯化抗体的稀释度为1∶3 200时,纯化抗体与PLRV仍呈现阳性反应(表1)。用PVS阳性标准物包被反应孔,间接ELISA测定显示,重组双CP多克隆抗体与PVS也呈现特异性反应,纯化抗体稀释度为1∶3 200时,纯化的抗体与PVS也呈现阳性反应(表2)。结果表明,重组双CP多克隆抗体与PLRV、PVS结合的特异性强、反应活性高。

M.蛋白分子量标准;1.重组双CP抗血清; 2.分离的抗体;3. 纯化的抗体。 M.Protein Marker;1.Antiserum against recombinant double CP; 2.Isolated IgG;3.Purified IgG.
在降低抗体稀释度(1∶500)与酶标二抗稀释度(1∶1 000)的情况下,用6种马铃薯主要病毒阳性标准物为抗原,间接ELISA反应显示,包被PVS或PLRV的反应孔呈颜色反应,包被其他4种病毒的反应孔无颜色反应(图5),并且OD值测定结果与目测结果一致。结果表明,本研究制备的重组双CP多克隆抗体只与PLRV、PVS有特异性反应,与其他4种病毒无交叉反应,能够满足ELISA检测的基本要求。

表1 PLRV与PVS重组双CP多克隆抗体(IgG)活性的间接ELISA测定(PLRV阳性物,Tab.1 Activity determination of IgG against double CP of PLRV and PVS by indirect

表2 PLRV与PVS重组双CP多克隆抗体(IgG)活性的间接ELISA测定(PVS阳性物,
综合重组双CP多克隆抗体特异性与活性测定结果,该抗体可用于PLRV的间接ELISA测定,也可用于PVS的间接ELISA测定,即同一种抗体可以用于PLRV与PVS两种病毒的间接ELISA检测。
2.3 PLRV与PVS的DAS-ELISA检测
为了进行DAS-ELISA反应,先用碱性磷酸酶标记纯化后的重组双CP多克隆抗体,直接ELISA测定表明,当用PLRV阳性标准物包被反应孔且酶标抗体的稀释度为1∶800,呈现阳性反应(表3);当PVS病毒标准物包被反应孔,酶标抗体的稀释度为1∶800,二者也呈现阳性反应(表4)。结果表明,酶标抗体与PLRV或PVS的结合特异性强、反应活性高。

表3 PLRV与PVS重组双CP抗体碱性磷酸酶标记物活性的直接ELISA测定(PLRV阳性物,

表4 PLRV与PVS重组双CP抗体碱性磷酸酶标记物活性的直接ELISA测定(PVS阳性物,
将制备的重组双CP多克隆抗体及其酶标抗体用于DAS-ELISA检测,其中重组CP多克隆抗体采用1∶100、1∶200与1∶300 3个稀释度,酶标抗体稀释度按照1∶100。结果表明,阴性对照及空白对照孔无颜色反应,加入PLRV或PVS的反应孔及阳性对照孔有颜色反应。OD测定结果表明,重组双CP多克隆抗体在3个稀释度下,以重组双CP为抗原的阳性对照反应孔中都呈现阳性反应(表5);当双CP多克隆抗体及其酶标抗体稀释度都为1∶100,PLRV反应孔呈现阳性反应,PVS反应孔也呈现阳性反应(表5)。结果表明,来自本研究制备的同一种抗血清的重组双CP多克隆抗体及其酶标抗体可用于PLRV的DAS-ELISA法的检测,也可用于PVS的DAS-ELISA检验,即一种抗体可检测2种病毒,这为提高马铃薯脱毒试管苗及种薯中病毒ELISA检测的效率及降低检测成本奠定了基础。

表5 PLRV与PVS阳性标准物的DAS-ELISA检测Tab.5 DAS-ELISA detection of PLRV and PVS positive
3 讨论
马铃薯病毒粒体分离纯化困难导致了马铃薯病毒特异性抗血清制备的困难,这限制了我国马铃薯病毒抗血清的大量制备及其在ELISA检测中的应用。利用重组CP作抗原便成为马铃薯病毒特异性抗血清(抗体)制备的一条重要的技术途径。除了上述PLRV与PVS外,国内外陆续针对PVX、PVY重组CP多克隆抗体的制备研究很多,可有效地用于间接ELISA测定,只有Balogun 等[10]发表了PVX重组CP多克隆抗体顺利地用于DAS-ELISA测定的结果及Jeevalatha等[11]发表了PVY重组CP多克隆抗体可用于PVY的DAS-ELISA检测的结果。目前,针对PVA及PVM的研究在逐渐增多[11-14],但PVA与PVM重组CP多克隆抗体制备的研究也局限于间接ELISA检测[15-17]。达到DAS-ELISA检测的要求,对于重组CP多克隆抗体在马铃薯病毒的ELISA检测中的应用至关重要。
本研究室进行马铃薯重组CP多克隆抗体制备研究与应用已经多年,针对6种马铃薯主要病毒,利用单CP制备的抗体都能达到DAS-ELISA检测的要求,目前只发表了PLRV与PVA[9]多抗的制备与应用结果。本研究涉及重组双CP抗体的制备及其应用,是在PLRV与PVS单CP多抗及其酶标抗体都达到DAS-ELISA测定要求的基础上进行的。前期研究中用溶菌酶裂解菌体,利用试剂盒推荐的蛋白纯化条件,都可以顺利地获得高纯度重组CP,本研究采用了融合基因,表达的蛋白产物分子量变大,纯化难度明显增大,主要表现为包涵体蛋白溶解困难,纯化产物含有杂蛋白,重组双CP溶解性降低很可能是肽链折叠方式不正确导致的。改用超声破碎法裂解菌体,得到的包涵体可溶性明显增加,溶解包涵体获得的蛋白溶解液中目的蛋白浓度及其相对含量都有明显提高,用同样的纯化条件就可获得高纯度重组双CP;此法中提取影响到纯化,实际上建立了获得高纯度重组双CP的提取与纯化一体化技术。同时制备的重组CP多克隆抗体可用PLRV及PVS为2种病毒的间接ELISA检测,也可用于DAS-ELISA检测,为进一步提高脱毒种薯病毒ELISA检测效率及降低检测成本奠定了基础。目前,用重组双CP制备植物病毒多抗的报道还很少,2014年,Kapoor等[18]制备出了黄瓜花叶病毒(CMV)与番木瓜环斑病毒(PRV)重组双CP多克隆抗体,可用于上述2种病毒的间接ELISA测定。同年,Kapoor等[18]还把截短的PVX与PVY融合双CP基因进行原核表达获得了重组双CP,制备的抗血清(抗体)可用于大田采集材料的间接ELISA检测[19]。
将单克隆抗体用于植物病毒ELISA检测是该检测技术的发展,单克隆抗体已经成功地用于PVS与PVM的ELISA检测[20-21],这是一个重要的发展方向。同时针对ELISA测定,特别是DAS-ELISA测定方法的改进一直不断,Rettcher等[22]用磁珠标记抗体,将DAS-ELISA原理用于PVX快速的磁侧向免疫层析检测,Panferova等[23]利用酪胺扩增酶反应信号,提高了ELISA反应的灵敏度,PVX的检出量从100 ng/mL降低到3 ng/mL。梁雨欣等[24]将酶标抗体与抗原结合后再加入包被抗体的反应孔中,建立了检测PVY的快速DAS-ELISA方法。作为植物病毒检测方法,ELISA检测仍呈现着巨大的应用活力。
猜你喜欢
环球时报(2022-09-20)2022-09-20
中国临床新医学(2022年8期)2022-09-08
科技视界(2022年10期)2022-05-20
健康之家(2021年2期)2021-07-01
昆明医科大学报(2019年2期)2019-09-10
祝您健康(2018年12期)2018-11-27
分析化学(2018年12期)2018-01-22
小资CHIC!ELEGANCE(2015年14期)2015-09-23
小资CHIC!ELEGANCE(2015年15期)2015-09-01
时代英语·高二(2015年2期)2015-05-18
