
对氨基苯酚印迹聚合物磁微粒制备及其萃取应用
2022-12-01 03:06褚琪琪王筱倩党雪平陈怀侠
湖北大学学报(自然科学版) 2022年3期
褚琪琪,王筱倩,党雪平,陈怀侠
(湖北大学化学化工学院, 湖北 武汉 430062)
0 引言
对氨基苯酚(p-aminophenol,PAP)和间氨基苯酚(m-aminophenol,MAP)常被应用于氧化型染发剂,但这些化合物可经皮吸收,长期使用会导致接触性皮炎或过敏,甚至会致癌、致畸[1]. 2015年,国家化妆品安全技术规范(STSC)明确规定氧化型染发剂中氨基苯酚类最大含量不允许超过0.5%[2].
已有多种方法用于测定氨基苯酚,如高效液相色谱法[3]、拉曼光谱检测法[4]、电化学检测法[5]和毛细管电泳法[6]等. 其中,高效液相色谱法由于操作方便、检测费用低而得到广泛应用.
染发剂样品基质复杂,干扰严重. 合适的样品前处理是其定量分析的关键,常用的方法有固相微萃取(SPME)、液相微萃取(LPME)和磁固相萃取(MSPE)等[7-9]. 磁固相萃取是固相萃取的新技术,具有快速分离的优点. 分子印迹聚合物具有分子识别选择性,是理想的吸附材料. 分子印迹磁材料兼具磁微粒和分子印迹的优势,在磁萃取中得到广泛应用,如应用于食品分析、环境分析、药物分析等样品前处理[10-12]. 目前,未见分子印迹磁材料应用于染发剂中氨基苯酚类化合物分离分析的研究报道.
本研究以共沉淀法制备出粒度均匀、分散良好、磁响应迅速的纳米Fe3O4,依次用正硅酸乙酯和3-(甲基丙烯酰氧)丙基三甲氧基硅烷对其进行表面修饰,采用表面印迹方法合成了对氨基苯酚的磁性分子印迹聚合物.以红外吸收光谱、X线衍射光谱、磁滞回线和热重分析进行材料的理化性能表征. 将该分子印迹磁性聚合物应用于染发剂中氨基苯酚类化合物的磁固相萃取,通过优化萃取条件,建立了染发剂中PAP和MAP的磁固相萃取-高效液相色谱定量分析方法,该方法操作简便、分离快速且选择性高.
1 实验部分
1.1 仪器与试剂LC-CT310型高效液相色谱仪(江苏天瑞公司);DAD230二极管阵列检测器(江苏天瑞公司);SMT-C18(4.6 mm×250 mm,5 μm)色谱柱(美国 Supelco Technologies);DZ-2BCⅡ型真空干燥箱(天津市泰斯特仪器有限公司);UPW-20 N 超纯水机(北京历元电子仪器有限公司); DW-Z型多功能化搅拌器(巩义市予华仪器有限公司);HH-2型数显恒温水浴锅(国华电器有限公司);WH-3 微型旋涡混合仪(上海沪西分析仪器厂有限公司);KQ 3200E型超声波清洗器(昆山市超声仪器有限公司); CP213 分析天平(上海五相仪器有限公司).
对氨基苯酚(PAP)、间氨基苯酚(MAP)、氯化铁(FeCl3·6H2O)、3-(异丁烯酰氧)丙基三甲氧基硅烷、2,2-偶氮二异丁腈(AIBN)、乙二醇二甲基丙烯酸甲酯和甲基丙烯酸(MAA)均为分析纯,购买于上海阿拉丁试剂有限公司;氯化亚铁(FeCl2·4H2O)、正硅酸乙酯、氢氧化钠、氨水(25%~28%)、无水乙醇、乙腈、醋酸和甲苯均为分析纯购买于上海国药集团化学试剂公司;醋酸胺购买于上海麦克林试剂有限公司;氯化钠为分析纯购买于天津博迪化工股份有限公司;甲醇色谱纯购买于美国Fisher公司.
1.2 色谱条件流动相组成为1 mmol/L的乙酸铵溶液∶甲醇(20∶80,V/V);流动相流速: 1.0 mL/ min; 色谱柱柱温:30 ℃;样品进样量:20 μL;测试波长:285 nm和300 nm.
1.3 磁性分子印迹聚合物的合成纳米Fe3O4的合成:4.053 g FeCl3·6H2O和1.987 5 g FeCl2·4H2O溶于80 mL水,在氮气保护下,以300 r/min的速度搅拌,逐滴加入50 mL氨水,80 ℃反应30 min,合成的Fe3O4微粒用超纯水洗涤多次,产物放入真空干燥箱,60 ℃真空干燥24 h.
Fe3O4@SiO2的合成:将0.3 g纳米Fe3O4,50 mL乙醇和4 mL超纯水进行混合,超声15 min,加入5 mL氨水、2 mL TEOS后,室温下反应12 h,产物用磁铁分离,经超纯水和乙醇洗涤至中性,放入真空干燥箱,60 ℃真空干燥24 h.
Fe3O4@SiO2@MPS的合成:将0.15 g Fe3O4@SiO2溶于30 mL无水甲苯,加入1 mL MPS,60 ℃水浴加热反应5 h,用磁铁分离产物,以乙醇洗涤,放入真空干燥箱,60 ℃真空干燥24 h.
磁性分子印迹聚合物(MMIPs)的合成:将0.5 mmol的对氨基苯酚溶于由10 mL甲醇、2 mL乙腈、5 mL氨水和3 mL 5 mmol/L乙酸铵溶液构成的混合溶液,加入170 μL MAA,超声30 min,放入4 ℃的冰箱中低温反应12 h.加入0.1 g Fe3O4@SiO2@MPS,搅拌并超声30 min,然后加入1.9 mL EGDMA和0.06 g AIBN, 在氮气保护下搅拌超声30 min,60 ℃水浴加热反应24 h.产物用磁铁分离,并用乙腈和乙醇洗涤数次,放入真空干燥箱,60 ℃真空干燥24 h.
磁性非印迹聚合物(MNIPs)的合成:除不加模板分子对氨基苯酚外,其他步骤同上.
1.4 标准溶液和样品溶液的配制标准溶液的配制:分别配制1 mg/mL PAP和MAP的甲醇储备液,存于冰箱4 ℃待用.实验前分别用超纯水稀释成所需浓度.
样品溶液的制备:将40 g染发剂样品溶于200 mL甲醇,超声15 min溶解,沉降2 h. 取上清液旋转蒸发溶剂,残留物用超纯水溶解并过滤,滤液转移至500 mL容量瓶并用超纯水定容,存于冰箱4 ℃待用.
1.5 磁萃取过程取10 mg分子印迹磁材料于培养瓶中,加入10 mL 200 ng/mL PAP和MAP的混合标准水溶液,震荡30 min,磁铁分离,除去上清液,用1 mL 1 mmol /L乙酸铵溶液∶甲醇(20∶80,V/V)混合液超声洗脱20 min,洗脱液过0.22 μm滤膜,过膜液直接进行高效液相色谱分析.
磁性分子印迹聚合物的制备流程和磁萃取过程见图1.
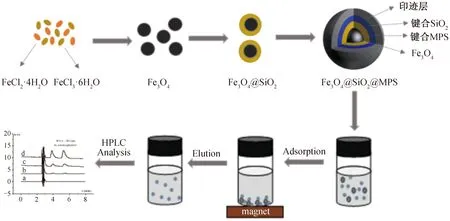
图1 对氨基苯酚印迹磁微粒制备流程和磁萃取过程图
2 结果与讨论
2.1 材料表征

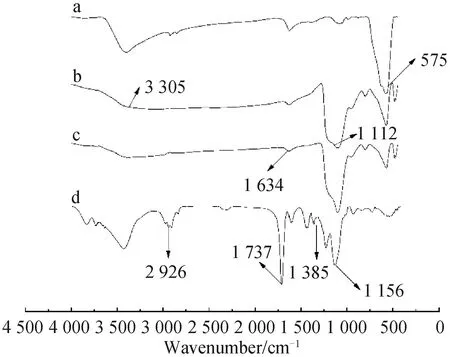
图2 Fe3O4(a)、Fe3O4@SiO2(b)、Fe3O4@SiO2@MPS(c)和对氨基苯酚印迹聚合物磁微粒(d)的红外光谱
2.1.2 XRD图谱分析 图3是Fe3O4(a)、Fe3O4@SiO2(b)、Fe3O4@SiO2@MPS(c)和对氨基苯酚印迹聚合物磁微粒(d)的X线衍射图,可见,Fe3O4的6个特征衍射峰出现在2θ=30.45°, 35.53°, 43.21°, 53.47°, 57.06° 和62.68°处,这同样与面心立方标准XRD图谱中Fe3O4的标准衍射峰对应,且没有明显的杂峰,说明Fe3O4纯度满足要求.而Fe3O4@SiO2、Fe3O4@SiO2@MPS和对氨基苯酚印迹聚合物磁微粒的主要衍射峰与Fe3O4的特征衍射峰位置一致,说明经表面修饰和印迹聚合物包覆后,Fe3O4的晶体结构未被破坏,该复合材料依然保持着良好的磁性.
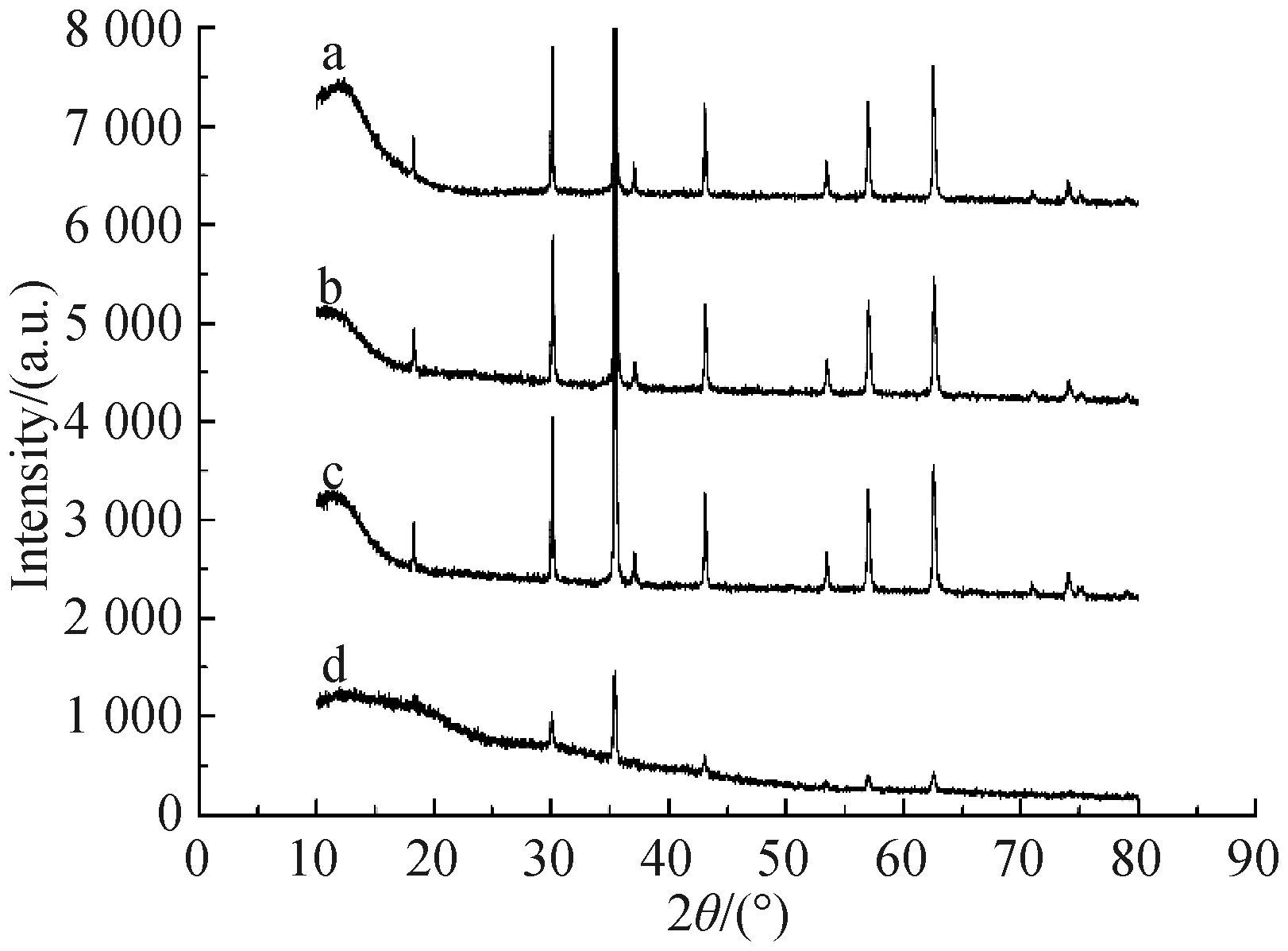
图3 Fe3O4(a)、Fe3O4@SiO2(b)、Fe3O4@SiO2@MPS(c)和对氨基苯酚印迹聚合物磁微粒(d)的X线衍射图
2.1.3 磁滞回线分析 Fe3O4(a)、Fe3O4@SiO2(b)、Fe3O4@SiO2@MPS(c)和对氨基苯酚印迹聚合物磁微粒(d)的磁滞回线分析图如图4所示.复合材料的饱和磁化强度值分别为69.62、55.44、48.93和4.17 emu/g,饱和值的下降表明表面修饰后,磁芯外表面SiO2、乙烯基和印迹层均键合成功,且仍然保持良好的磁性,在外加磁场条件下,能满足快速分离的要求.
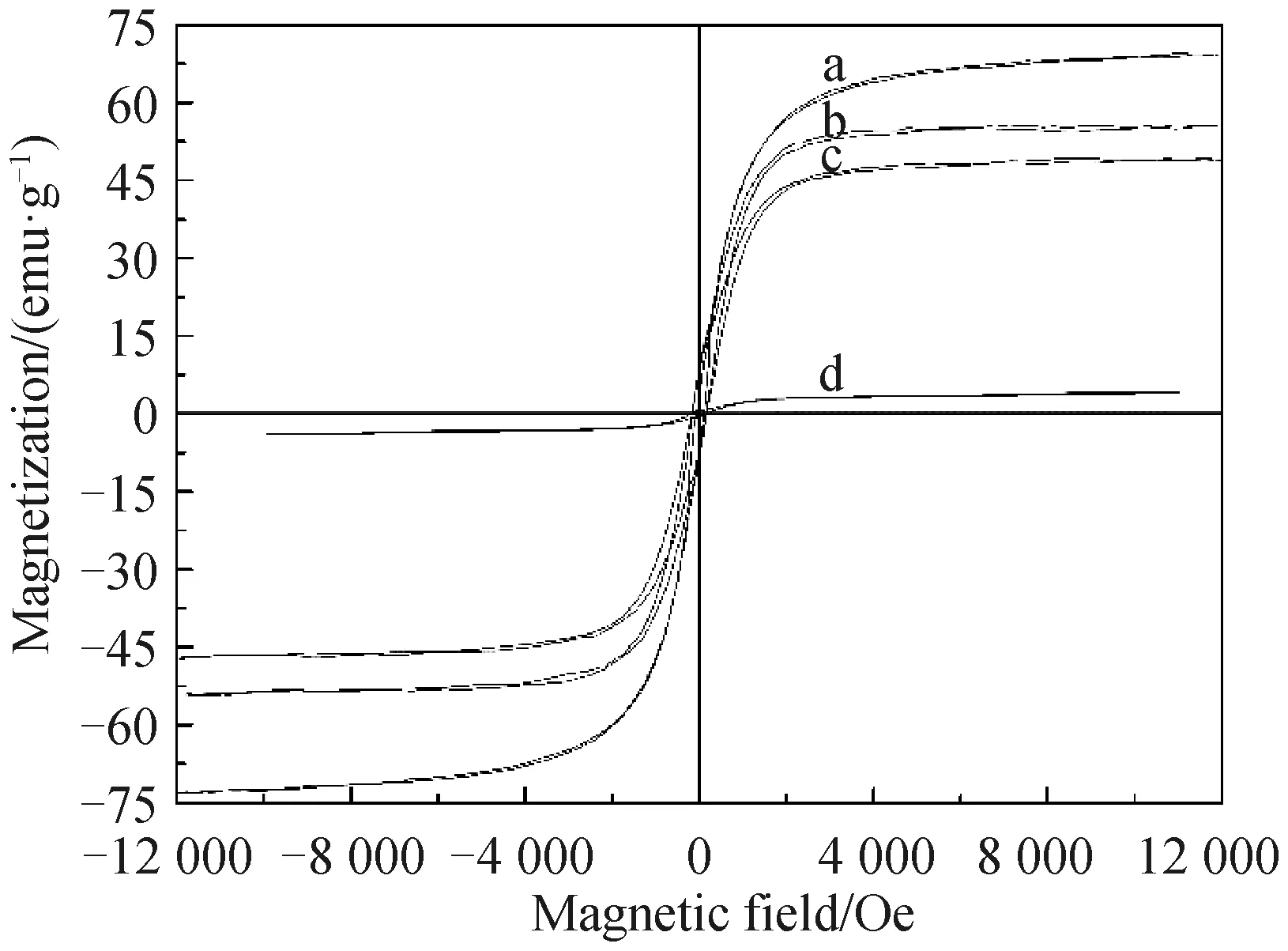
图4 Fe3O4(a)、Fe3O4@SiO2(b)、Fe3O4@SiO2@MPS(c)和对氨基苯酚印迹聚合物磁微粒(d)的磁滞回线分析图
2.1.4 热重分析 图5是对氨基苯酚印迹聚合物磁微粒的热重分析曲线.可见,合成的印迹磁微粒在300 ℃以下具有良好的稳定性.
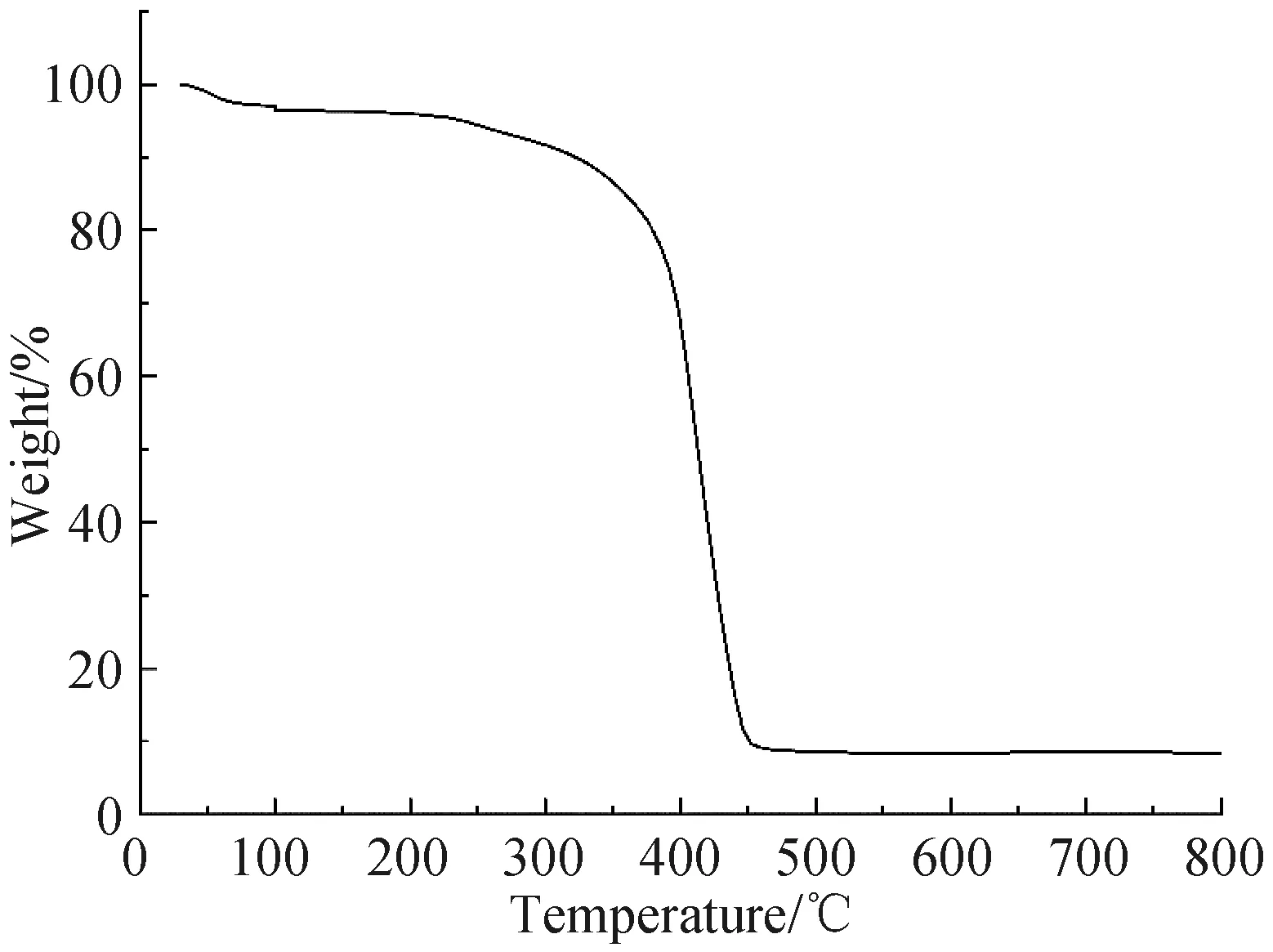
图5 对氨基苯酚印迹聚合物磁微粒热重分析图
2.2 印迹因子选择性以富集倍数(EF)和印迹因子(IF)进行评价,两者的定义为:
式中C0为样品溶液分析物的浓度,Celu为洗脱液中分析物浓度,EFMMIP为印迹物对分析物的富集倍数,EFMNIP为非印迹物对分析物的富集倍数.
如表1所示,该印迹材料对PAP和MAP的印迹因子IF分别为2.56和2.27,说明该材料对PAP和MAP具有良好的吸附选择性,适用于实际样品中的氨基苯酚类化合物的分离萃取.

表1 MMIPs和MNIPs对对氨基苯酚和间氨基苯酚的富集倍数和印迹因子
2.3 萃取条件的优化
2.3.1 上样时间的考察 在相同萃取条件下,取5份10 mL 200 ng/mL对氨基苯酚和间氨基苯酚混标溶液进行萃取,考察上样时间分别为10、20、30、40、50 min时的回收率.结果显示,上样时间30 min时的吸附效果最好.因此,后续实验中的上样时间为30 min.
2.3.2 洗脱时间的考察 在相同的萃取条件下,考察洗脱时间分别为5、10、15、20和25 min时的回收率.结果显示,洗脱时间为20 min时,具有良好的洗脱效果.因此,后续实验选择洗脱20 min.
2.3.3 上样体积的考察 在相同萃取条件下,分别取10、20、30、40和50 mL 200 ng/mL分析物混标溶液上样,考察样品体积对于回收率的影响.结果表明,10 mL样品上样时,目标物的回收率最高.因此,后续实验选择10 mL样品溶液上样.
2.3.4 洗脱剂组成的考察 在相同萃取条件下,以1 mmol/L乙酸铵溶液:甲醇分别为10∶90、20∶80、30∶70、40∶60和50∶50 (V/V)的混合液为洗脱剂,考察洗脱剂组成对回收率的影响.实验结果显示,乙酸铵溶液和甲醇的体积比为20∶80时,目标物的回收率最高.因此,后续实验选择1 mmol/L乙酸铵溶液∶甲醇=20∶80 (V/V)的混合液为洗脱液.
2.3.5 洗脱剂体积的考察 在相同萃取条件下,分别用0.5、1.0、1.5、2.0和2.5 mL洗脱液进行洗脱.结果显示,1.0 mL洗脱液能够实现定量洗脱,综合考虑上样体积和富集倍数,后续实验选择1 mL洗脱液.
2.3.6 溶液酸度的考察 在相同萃取条件下,在pH为6~11范围内考察溶液酸度对萃取率的影响.结果显示,样品溶液pH值从6增加到7时,分析物的回收率呈现上升趋势,pH 在7~9时,回收率基本不变,而pH>9时,回收率再次下降.这是因为氨基苯酚的两级解离常数(pKa)分别为5.43和10.40,即在pH<6.43的酸性条件下,氨基苯酚质子化为阳离子,在pH>9.40的碱性条件下,氨基苯酚会解离为阴离子,都不利于印迹材料的吸附分离.只有在pH约为7~9时,氨基苯酚以分子状态存在,印迹材料的分子识别吸附使得回收率较高.即材料的印迹和分子识别吸附主要是氢键作用.样品溶液的pH值约为8,因此,在后续实验中,不改变溶液pH值.
2.3.7 溶液盐度的考察 其他萃取条件不变,为考察溶液中离子强度对萃取效率的影响,在溶液中加入不同量的NaCl固体(0~20%,w/v).实验结果显示,样品溶液中加入NaCl后,目标物的回收率明显下降.因此,在后续实验中,样品溶液中不加盐.
2.4 方法评价在优化的萃取条件下,建立染发剂样品中对氨基苯酚和间氨基苯酚的MSPE-HPLC定量分析方法. 如表2所示,以Y表示峰面积,X表示分析物的浓度,对氨基苯酚在0.06~50 mg/kg线性范围内线性关系良好,线性回归方程为Y=25.672 0X-1.135 6 (R2=0.998 9);间氨基苯酚在0.08~50 mg/kg线性范围内线性关系良好,线性回归方程为Y=27.251 0X-0.492 1(R2=0.998 3).检出限(LOD,S/N=3)分别为0.018 mg/kg和0.024 mg/kg;定量限(LOQ,S/N=10)分别为0.060 mg/kg和0.080 mg/kg.
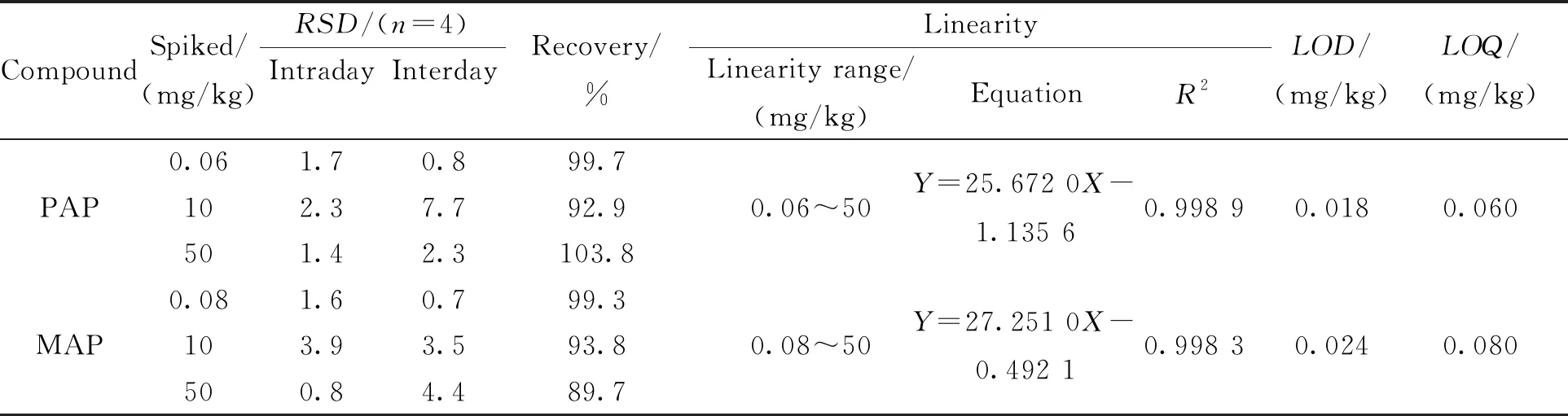
表2 线性范围、检测限和定量限
为考察方法的准确度和重现性,在空白染发剂中分别加入0.06、10和50 mg/kg对氨基苯酚和间氨基苯酚. 如表2所示,目标物的加标回收率在89.7~103.8 %范围内,日内和日间精密度(RSD)分别为0.8%~3.9%和0.7%~7.7%,说明该方法准确度高、重现性好,能满足染发剂样品中氨基萘酚类化合物的定量分析.
2.5 实际样品分析将本方法应用于超市购买的两种染发剂检测.按照1.4制备样品溶液,结果发现,两种样品中对氨基苯酚的含量分别为0.46%和1.02%,间氨基苯酚的含量分别为为0.37%和0.78%.
2.6 方法比较将本方法与其他氨基苯酚的分析方法进行比较,如表3所示.可见,本方法比现有方法的检出限低很多,线性范围更宽,即分子印迹材料对目标物具有良好的分离富集效果.本方法适用于染发剂样品中氨基苯酚类化合物的分离分析.
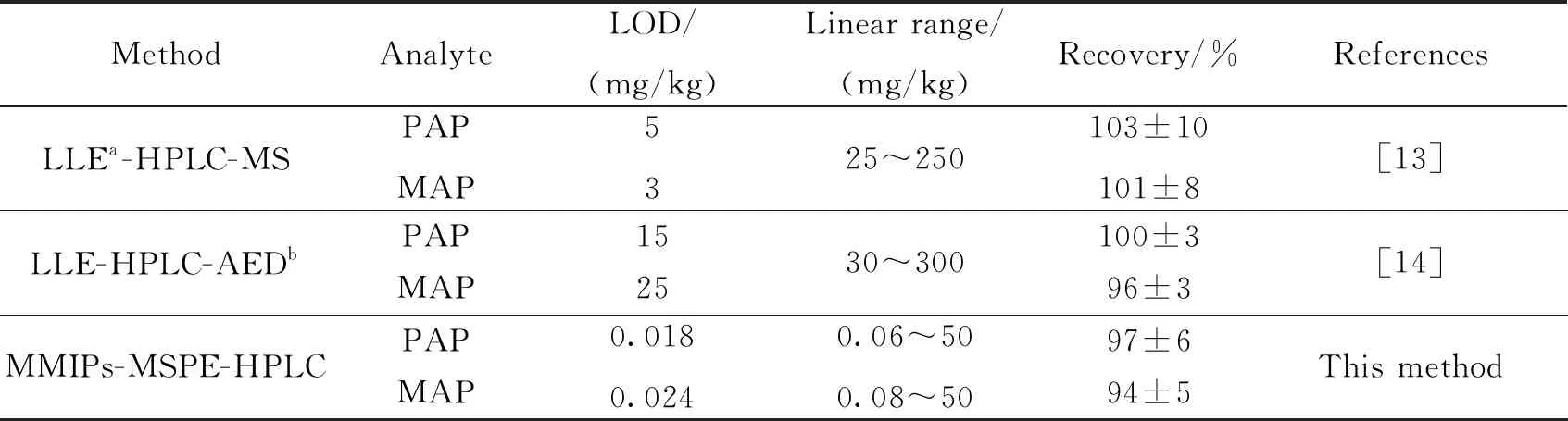
表3 本方法与其他方法的比较
3 结论
在SiO2和MPS双重修饰的纳米Fe3O4表面成功合成了对氨基苯酚分子印迹聚合物.该材料具有良好的热稳定性和磁性,对于氨基苯酚类化合物具有良好的分子识别吸附选择性,应用于磁固相萃取,该萃取方法简便快速,分离富集效果好.建立了染发剂中氨基苯酚类化合物的MSPE-HPLC分析方法. 该方法选择性好、灵敏度高、重现性好,且操作简便,适用于染发剂样品中氨基苯酚类的定量分离分析.
猜你喜欢
国企管理(2022年3期)2022-05-17
云南画报(2021年10期)2021-11-24
家庭医学(2021年9期)2021-09-28
家庭科学·新健康(2021年3期)2021-04-08
保健与生活(2021年6期)2021-03-16
大众健康(2021年2期)2021-03-09
师道·教研(2019年10期)2019-11-21
小学生优秀作文(高年级)(2018年4期)2018-09-11
学校教育研究(2017年28期)2017-10-21
科技创新与应用(2017年20期)2017-07-15
