
牡蛎相关微病毒科噬菌体基因组的鉴定和进化分析
2023-10-24 03:18刘广锋谢科明姜敬哲
南方水产科学 2023年5期
刘 敏,刘 畅,刘广锋,朱 鹏,谢科明,姜敬哲
1.上海海洋大学 海洋生态与环境学院,上海 201306
2.中国水产科学研究院南海水产研究所/农业农村部南海渔业资源开发利用重点实验室,广东 广州 510300
3.广东药科大学 生命科学与生物制药学院,广东 广州 510005
牡蛎 (Crassostrea) 是一种双壳类软体动物,广泛分布于世界各地沿岸潮间带,其肉质鲜美、营养丰富,是世界第一大养殖贝类,也是我国支柱性海水养殖种类之一[1]。由于养殖环境恶化、养殖密度过高和种质退化等原因,病害问题已经成为制约牡蛎产业发展的重要因素。其中细菌性病原的危害尤其严重[2]。细菌病害的发生与养殖密度和水质环境密切相关,但由于牡蛎大多在开放海域养殖,直接施用消毒剂、抗生素等药物会被海水快速稀释,不但起不到防治作用,还会对海洋生态环境造成破坏。
噬菌体是专性侵染细菌的病毒,能高效、精准地攻击宿主细菌,在海洋生态系统中扮演着重要角色。使用噬菌体治疗细菌性疾病已在医疗[3]、畜禽[4]和水产[5]领域得到了广泛实践,是减抗替抗的重要选择。虽然细菌变异很快,但噬菌体拥有高度多样化的基因库和极高的变异速度[6],能快速获得新功能从而适应宿主及环境变化[7-8],实现控制宿主细菌丰度、微生物群落组成,并影响动物宿主健康,甚至对海洋生物地球化学循环产生影响[9]。迄今为止,对具有双链DNA (double strands DNA,dsDNA) 基因组的噬菌体的研究较为充分,已经有大量关于它们的多样性、进化及在细菌性病害治疗方面的研究报道。然而,针对单链DNA (single strand DNA,ssDNA) 噬菌体的研究仍然非常缺乏。与dsDNA 噬菌体相比,ssDNA 噬菌体的基因组较小,且在各种环境中广泛存在[10],便于直接人工合成,通常不携带毒力或抗性基因等有害基因,是进行功能噬菌体开发的理想类群。
本课题组的前期研究发现,养殖牡蛎体内蕴藏着异常丰富、多样且全新的病毒和噬菌体类群,其中大量与微病毒科 (Microviridae) 和圆环病毒科(Circoviridae) 等ssDNA 病毒相关[11]。微病毒科噬菌体是一类具有环状ssDNA 基因组的噬菌体[12],在水环境、动物和人类内脏的内容物中均有检出[13-15]。微病毒科包含两个经由国际病毒分类委员会 (International Committee of Taxonomy of Viruses,ICTV) 批准的亚科:Bullavirinae和Gokushovirinae,两个亚科之间的区别主要在于基因组组成和主要衣壳蛋白的结构不同。Bullavirinae以微病毒PhiX174 为代表,包括Alphatrevirus、Gequatrovirus和Sinsheimervirus3 个属,据报道,该亚科可感染肠杆菌属 (Enterobacter) 细菌,并已通过其代表病毒株PhiX174 进行了深入的分子学研究[16]。Gokushovirinae亚科因其可感染细胞内寄生生物衣原体属 (Chlamydia)、螺原体属(Spiroplasma) 和蛭弧菌属 (Bdellovibrio)[12],通常被称为“寄生虫的寄生虫”。
近年来,在多种宏基因组样本中也发现了大量未分类的微病毒科噬菌体,或许会进一步扩充微病毒的宿主范围[13-15]。本研究对华南沿海多地养殖牡蛎样本的宏病毒组测序数据进行深入挖掘,发现并鉴定到5 个感染埃希氏菌属 (Escherichia) 的新型微病毒科噬菌体,相关成果可为推动微病毒科噬菌体多样性与分类研究、研发具有良好抑菌作用的新型ssDNA 噬菌体制剂提供参考。
1 材料与方法
1.1 数据来源
本课题组已完成华南多地养殖牡蛎样品的宏病毒组测序工作,共获得了约25 亿条测序原始序列(reads)[11]。使用 Fastp (V0.20.0)[17]对原始序列进行质控,包括去除低质量和接头序列,然后使用Megahit (V1.2.9)[18]将过滤后的reads 组装成重叠群(Contig),进一步使用Diamond (V0.9.24.125)[19]比对美国国家生物技术信息中心 (National Center for Biotechnology Information,NCBI) 的非冗余蛋白质数据库 (NR),从而对Contig 序列进行注释。使用Megan 6[20]对注释结果进行进一步分类鉴定,选择其中注释为微病毒科的病毒序列,经病毒基因组完整性鉴定后共获得了227 个几乎完整的疑似微病毒基因组。
1.2 微病毒序列筛选
选用微病毒科中经ICTV 分类的8 个属 (分属于两个亚科) 的15 条序列作为参考株,与新获得的227 条微病毒序列一并分析。使用Prodigal(V2.6.3)[21]预测开放阅读框 (Open reading frame,ORF),run type 选择normal mode,mode 选取meta。使用Diamond (V0.9.24.125) 对预测的ORF 进行Blastp 比对,比对数据库为NCBI 非冗余蛋白质数据库。筛选出其中比对结果为主要衣壳蛋白且相似度最高的前10 条序列,除去重复序列后,共得到741 条相似度较高的主要衣壳蛋白序列。使用gephi (V0.9.2)[22]绘制主要衣壳蛋白的网络关系图,选取与Bullavirinae聚类的5 条序列进行深入分析 (Contig ID: HSd1-5344568、GX1-198598、T4S1-22425、T4S1-854210、ML1-11067)。
1.3 主要衣壳蛋白进化树分析
使用MAFFT (V7.515)[23]将所选取的序列进行比对,并使用TrimAl[24]对结果进行裁剪。采用iqtree (V2.2.0.3)[25]进行最大似然系统发育分析,设置为自动选择最优模型,Bootstrap 为1 000,最后使用itol (https://itol.embl.de/) 可视化树文件。
1.4 基因组结构分析
使用Orthologous Average Nucleotide Identity Tool (OAT) (V0.93.1)[26]计算5 条病毒序列与其在进化树上的相邻序列之间的平均核苷酸相似性(Average Nucleotide Identity,ANI)。使用Cenote Taker2 (V2.1.3)[27]对有代表性的若干基因组的ORF进行功能注释,并使用SnapGene (V4.3.6,www.snapgene.com) 用于打开Cenote-Taker2 输出文件,将基因组结构可视化,展示在进化树中对应的序列之后。
1.5 宿主预测
使用CHERRY (V0.9.24.125)[28]对进化树上的组装病毒序列进行宿主预测。CHERRY 集成了多种基于蛋白质和 DNA 的序列特征 (如噬菌体之间的蛋白质信息、噬菌体与原核生物之间的序列相似性、CRISPR 信号以及k-mer 频率信息),利用这些特征计算配对病毒和原核生物之间形成感染的概率,选择概率最高的为预测的细菌宿主。
1.6 病毒丰度分析
为了计算每个病毒的相对丰度值,首先用salmon (V0.13.1) index[29]命令对合并后的5 条病毒基因组序列构建索引 (Index),作为salmon 比对的参考基因组数据集;然后再利用salmon quant[29]命令将牡蛎病毒组文库全部的clean reads 与参考基因组进行映射 (Mapping),统计能映射到这5 条基因组的reads 数量,根据调整后的TPM (Transcripts per million) 计算公式计算这5 条病毒序列的相对丰度值。
2 结果
2.1 牡蛎微病毒科相关病毒的发现与聚类
本研究在前期工作的基础上,以中国华南沿海多地养殖牡蛎为研究对象,进行宏病毒组测序后,去除低质量序列并组装,得到3 347 421 条非冗余的Contig 序列,并与NCBI 非冗余蛋白序列库进行blastp 比对和分类鉴定,最终获得728 784 条疑似病毒来源的Contig。由图1 可见,牡蛎样本中(橙色点) 发现了大量的疑似微病毒科噬菌体,其中绝大多数为全新的分类群,与NCBI 中未分类的相关噬菌体聚类,并与ICTV 参考毒株 (绿色点) 差异明显。由于未分类病毒类群的分析难度较高,本研究进一步筛选其中与Bullavirinae参考株序列聚类在一起的5 条疑似微小病毒的基因组序列 (图1,橙色圈) 进一步分析。
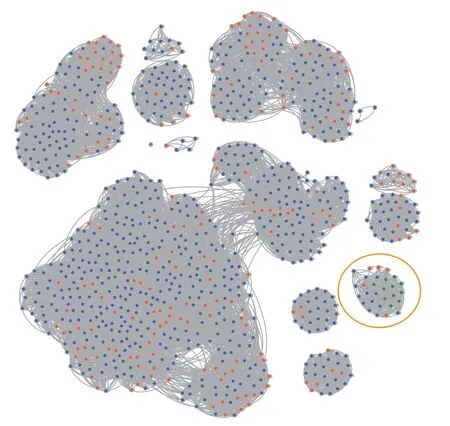
图1 主要衣壳蛋白的相似性网络聚类图注:图中的点表示主要衣壳蛋白序列,绿点代表ICTV 分类的微病毒科 (n=15),橙点表示牡蛎样本中鉴定为微病毒科的序列 (n=220),蓝点 (n=741) 表示上述两个来源的序列在nr 库中最相似的序列,灰线表示连接两个点的边 (即两点序列间blastp 打分值)。Fig.1 Identity network of major capsid proteinsNote: The dots in the figure indicate the major capsid protein sequences,among which the green dots (n=15) represent the Microviridae classified by ICTV; the orange dots (n=220) indicate sequences identified as Microviridae in oyster samples; the blue dots (n=741)indicate the sequences from the two sources mentioned above which are most similar in the NR library; the gray line indicates the edge connecting the two dots (Based on the score values of blastp).
2.2 微病毒科成员的系统发育、宿主来源与基因组结构
主要衣壳蛋白是微病毒科分类的经典系统发育标志基因[30]。为了解这5 条病毒序列的进化关系和分类界定,对5 条序列、与其相关的近缘病毒及经ICTV 认定的微病毒科参考株序列的主要衣壳蛋白序列汇总构建进化树 (图2)。进化树分成3 个大的分支,其中HSd1-5344568 与其他4 条序列聚类到不同的分支中,与已被报道的序列QBQ83335.1亲缘关系最近,聚集在亚科Bullavirinae的大分支。其余4 条序列聚类在Bullavirinae和Gokushovirinae两个亚科分支的中间分支中,其中T4S1-22425、T4S1-854210 和 GX1-198598 这3 条序列聚类在同一个小分支中,且没有其他已知的相似序列。利用Cherry 软件进行宿主预测的结果显示,Gokushovirinae分支的宿主主要为螺原体、蛭弧菌和衣原体属。Bullavirinae分支以及新发现的分支中大多数噬菌体的宿主为埃希氏菌属、肠杆菌属和乳球菌属 (Lactococcus) ,符合已报道的宿主范围,并且本研究的5 条噬菌体基因组均来自埃希氏菌属(棕色标注)。

图2 主要衣壳蛋白进化树及对应的基因组结构图注:MAFFT 比对主要衣壳蛋白的氨基酸序列,TrimAl 序列对齐,用iqtree 建树,itol 可视化,最大似然数;自展值:1 000,自动选择最佳替代模型,临界值70%。进化树上的红色星形表示来自牡蛎样品的微病毒科序列,序列号为:GX1-198598、T4S1-854210、T4S1-22425、ML1-11067 和HSd1-5344568;ID 的背景色表示用Cherry 预测的宿主类型;ID 右边一列表示ICTV 分类的亚科和属,以及未分类病毒的样品来源;右侧为基因组结构示意图,不同颜色表示不同的标志基因。Fig.2 Phylogenetic tree of major capsid proteins and related schematic maps of genome structureNote: The amino acid sequences of the major coat proteins were aligned by using MAFFT,trimmed by using trimAL sequences,phylogenetic tree was drawn by using iqtree,visualized by itol,Maximum Likelihood Estimation tree; bootstrap replication: 1 000,automatic selection of the best alternative model,and site coverage cutoff is 70%.The red ID on the phylogenetic tree indicates the sequence from the oyster sample.The background color of ID represents the host type predicted by Cherry; the column on the right side of ID shows the subfamilies and genera of ICTV classification,as well as the sample sources of unclassified viruses; and on the right side is a schematic map of genome structure,with different colors indicating different marker genes.
由样品来源看 (图2,中间注释信息),未知病毒数据主要来源于动物、废水和沉积物共3 类样品。但在5 个牡蛎病毒各自的进化分支上,同一分支的相邻序列也均来自于动物宏基因组样本,说明这些微病毒的宿主可能是在动物宿主体内专性共生的细菌。
根据基因组功能基因注释 (图2) 发现:首先,微病毒科噬菌体具有比较保守的基因组结构。主要衣壳蛋白 (黄色) 和复制相关蛋白 (粉色) 是其最具标志性的两个保守基因,两个基因并不相邻,中间会夹杂一些其他基因。其次,微病毒科的编码基因均为同一方向,即均为正义链编码,与圆环病毒等其他环状ssDNA 病毒不同。最后,微病毒科3 个主要亚科之间存在差异明显的基因组结构。位于主要衣壳蛋白和复制相关蛋白之间的次要衣壳蛋白(橙色) 是Gokushovirinae的特征基因;两种刺突蛋白 (绿色和深蓝色) 和外部支架蛋白 (浅蓝色) 是Bullavirinae的特征基因;而作为Bullavirinae姊妹分支的中间分支同样拥有外部支架蛋白,但刺突蛋白似乎并不是其特征基因。值得一提的是,T4S1-22425 基因组中未能预测到复制相关蛋白的基因,这或许说明该病毒拥有非常独特的复制相关蛋白,当然也无法排除因测序或组装导致的序列错误。本研究还发现,归属于Bullavirinae的HSd1-5344568与相似序列QBQ83335.1 的基因组结构较为一致,但不同于ICTV 标准Bullavirinae毒株,它们都缺少次要刺突蛋白。
2.3 平均核苷酸一致性
由图2 可知,GX1-198598、T4S1-22425 和T4S1-854210 3 条序列的主要衣壳蛋白聚类在一起,HSd1-5344568 与QBQ83335.1、ML1-11067 与AXL15490.1 分别在同一个聚类中。为了进一步比较几个近缘病毒之间的相似性,进一步分析了这些病毒基因组间的两两平均核苷酸相似性 (ANI)(图3)。GX1-198598、T4S1-22425 和T4S1-854210 3 个基因组之间的ANI 为0。HSd1-5344568 和ML1-11067与各自最相近序列的A N I 分别为6 6.2 3% 和62.09%。综合进化树的聚类和ANI 的结果,推测5 条序列中HSd1-5344568 属于Bullavirinae亚科的一个新属,其余4 条序列则属于一个新的亚科。ICTV 的分类标准判断,这5 条序列与任一参考微病毒科病毒序列的平均核苷酸相似性均低于70%,因此至少应该分别代表了5 个新的属。

图3 微病毒科基因组序列间的两两平均核苷酸相似性Fig.3 ANI value among genomic sequences
2.4 主要衣壳蛋白结构预测
通过结构预测结果,可以辅助判断证实蛋白的功能及活性。从本研究对5 个新型病毒的主要衣壳蛋白结构域三维结构预测结果可见 (图4),5 条序列的主要衣壳蛋白的空间结构存在较高的相似性,所有病毒的主要衣壳蛋白均具有保守的8 股β 桶核心 (也称为病毒果冻卷)。其中图4-a、4-b 和4-c为进化树上相邻的3 个分支,这3 个序列的三维结构也非常相似。HSd1-5344568 (图4-d)与其相似序列QBQ83335.1 (图4-e)、ML1-11067 (图4-f)与其相似序列AXL15490.1 (图4-g) 之间的空间结构相似性均较高。
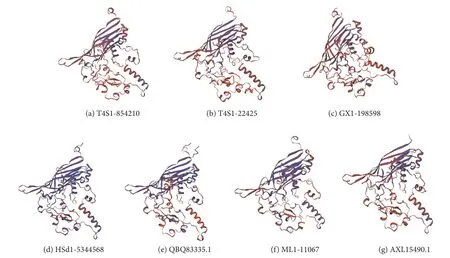
图4 微病毒科噬菌体主要衣壳蛋白三维结构预测图Fig.4 Three-dimensional structure prediction of replication proteins of oyster-related Microviridae
2.5 主要衣壳蛋白进化树与外部支架蛋白的进化规律与重组
为了更好地了解微病毒中各基因的进化规律,将主要衣壳蛋白进化树与外部支架蛋白的进化树进行了关联分析。由图5 所示,无论是外部支架蛋白还是主要衣壳蛋白,Gokushovirinae序列都聚集在一个独立的进化分支中,说明Gokushovirinae亚科的进化位置或生态位相对独立,未发现与其他亚科病毒的重组现象。另一方面,虽然外部支架蛋白进化树的总体结构与主要衣壳蛋白相似,即Gokushovirinae、Bullavirinae和中间分支相互独立(图5 中间黑色和蓝色连线),但发现其中有两个病毒 (NC001330 和KF044309) 的基因可能发生了重组(图5 中间红色连线),即重组了属于Bullavirinae的主要衣壳蛋白和属于中间分支的外部支架蛋白。值得注意的是,KF044309 与其姊妹分支KX266637 的宿主均为肠杆菌属 (图2),与临近的其他病毒宿主 (埃希氏菌属) 明显不同。该结果提示微病毒的中间分支类群与Bullavirinae类群可能在一定程度上存在宿主或生态位重叠,从而为两种病毒基因组的重组提供了机会。
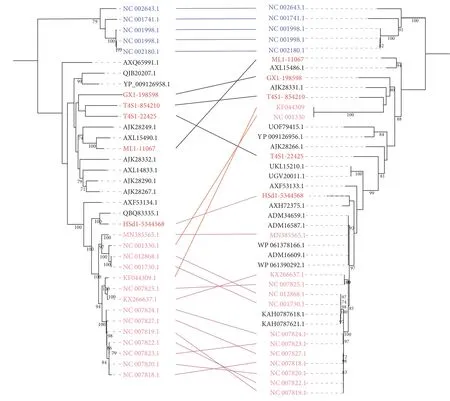
图5 微病毒科噬菌体主要衣壳蛋白与外部支架蛋白的进化关系注:使用 MAFFT 进行基因组序列比对,TrimAl 对齐氨基酸序列,iqtree 建树,itol 可视化,最大似然数;自展值:1 000,替代模型:自动选择。图中紫色序列ID 为Gokushovirinae 序列,红色序列ID 为牡蛎相关微病毒科序列,粉色序列ID 为Bullavirinae 序列。左边为主要衣壳蛋白的进化树,右边为外部支架蛋白的进化树,同一个基因组的两个蛋白用直线连接。Fig.5 Linkage between phylogenetic tree of major coat protein and that of external scaffolding protein of Microviridae phageNote: The amino acid sequences of the major coat proteins were aligned by using MAFFT,trimmed by using trimAL sequences,phylogenetic tree was drawn by using iqtree,visualized by itol,Maximum Likelihood Estimation; bootstrap replication: 1 000,automatic selection of the best alternative model,and site coverage cutoff value is 70%.The purple ID in the figure is Gokushovirinae; the red ID is the oyster-associated Microviridae sequence,and the pink ID is the Bullavirinae sequence.The phylogenetic tree of the major capsid protein is shown on the left,and the evolutionary tree of the external scaffold protein is shown on the right,with two proteins of the same genome connected by straight lines.
2.6 牡蛎相关微病毒的丰度
分析病毒序列在测序数据中的丰度可为了解病毒的生态功能提供线索,噬菌体的丰度与其细菌宿主的丰度密切相关。为了计算每个噬菌体序列的相对丰度,统计了Salmon 软件映射到5 条基因组上的reads 数量,其中只有除ML1-11067 外的4 条序列有映射结果。由TPM 丰度表 (表1) 可见,4 条序列在文库中的丰度均比较低,说明这些病毒的宿主在牡蛎体内的丰度并不高,对宿主的影响较小。HSd1-5344568 在样品中的丰度最高、分布最广。其中ML1-11067 在各个文库中都没有映射到reads,可能是组装软件Meghit 与映射软件Salmon 的算法不同所导致。

表1 牡蛎相关微病毒在各测序文库中的 TPM 丰度Table 1 TPM abundance of oyster-related microviruses in sequencing libraries
3 讨论
迄今为止,大多数水生生物的微生物组研究都集中在细菌群落上,其病毒组直到最近才受到关注[31]。噬菌体与微生物群落和动物宿主都有很强的相关性[32],在肠道等复杂的生态系统中,多种因素可能会影响噬菌体的丰度、多样性和生态作用。然而,由于噬菌体基因组中缺乏通用的基因标记,因此很难鉴定噬菌体的群落组成[33]。病毒宏基因组学对给定样本中的全体病毒核酸进行高通量测序,是探索新型病毒、挖掘病毒多样性的一种高效方法[15]。
最近的研究表明,人们已在多种环境中发现了众多微病毒科的成员,提示它们可能在不同的环境中发挥作用。如在不同自然栖息地或动物粪便和组织的宏基因组数据中,已经组装出数千个完整的微病毒基因组。这些环境微病毒都揭示了Gokushovirinae的巨大多样性,为将新发现的大量病毒进行合理分类,研究人员提出了一些假定亚科,其中包括从泥炭地生态系统中发现的Aravirinae和Stokavirinae[13]、从白蚁肠道中发现的Sukshmavirinae[34]、蜻蜓相关的D 类群[10]以及从人类肠道中发现的Alpavirinae[35]。本研究结果显示,5 条新型微病毒序列中,仅HSd1-5344568 被分类为Bullavirinae,其余4 条候选序列 (GX1-198598、T4S1-22425、T4S1-854210、ML1-11067) 虽然与ICTV认定的两个标准亚科没有聚类到一起,但明显属于介于这两个标准亚科的中间类群,与之前研究报道的其他假定类群不同。
近年来,养殖贝类的细菌性疾病连年暴发,严重阻碍了贝类养殖业的发展。目前的主流抗菌手段是使用抗生素,但其大规模且不规范的使用也带来了水产品药物残留、细菌耐药性和环境污染等突出问题,因此亟需寻求安全、天然无残留的抗生素替代品。噬菌体作为一种特异性高且生态友好的抗生素替代品引起了广泛关注,有实验表明噬菌体可以有效预防和控制水产养殖中的病原菌[32,36]。然而由于噬菌体具有宿主专一性,导致其抗菌谱较狭窄,但这一问题可以通过使用噬菌体鸡尾酒法来解决[37],即使用3 种或以上的噬菌体联合应用来杀灭细菌。所以在水生环境和水生动物样品中挖掘噬菌体的多样性对研发噬菌体抗菌制剂十分重要。
通过宿主预测软件的分析发现,这5 个噬菌体的宿主可能为埃希氏菌属细菌,与先前报道的Bullavirinae噬菌体的宿主范围一致。埃希氏菌属属于肠杆菌科,包括大肠埃希氏菌 (E.coli)、费格森埃希氏菌 (E.fergusonii) 等5 个种,其中致病性大肠埃希氏菌被认为是21 世纪对人类健康产生重大影响的病原微生物之一[12]。Bullavirinae亚科的噬菌体还可能感染乳球菌属 (Lactococcus) 和肠杆菌属。乳球菌属包括格氏乳球菌 (L.garvieae)、乳酸乳球菌 (L.lactis) 等种或亚种,其中格氏乳球菌和鱼乳球菌 (L.piscium) 已被报道可引起鱼类、中华鳖(Trionyxsinensis)、罗氏沼虾 (Macrobrachiumrosenbergii) 等水产动物发生腹水性肝脾病变、腹腔和内脏出血以及体表溃疡等病症,是危害淡水养殖的常见病原菌[38-39]。肠杆菌属也是肠杆菌科中的一员,广泛分布于自然环境中,目前已发现该属的14 个种,属于重要的机会性致病菌。根据中国细菌耐药网提供的数据,2015—2017 年肠杆菌属检出率均排在前十位,尤其是该菌属下的阴沟肠杆菌检出率均占所检出革兰氏阴性菌第五位。
综上所述,本研究围绕5 条与ICTV 标准病毒株近缘的新型微病毒的全基因组序列,通过对包括宿主来源、基因组结构、主要衣壳蛋白、外部支架蛋白等多方面的分析,明确了5 个微病毒的宿主来源及其分类位置,并发现了微病毒主要衣壳蛋白和外部支架蛋白存在的重组现象。本研究可为拓展海洋病毒多样性[40]、理解微病毒科病毒的分类进化规律、深入挖掘海洋噬菌体资源提供参考。
猜你喜欢
解放军医学院学报(2022年1期)2022-04-02
昆明医科大学学报(2022年2期)2022-03-29
天津市教科院学报(2021年5期)2021-11-10
中国动物传染病学报(2021年3期)2021-07-21
生物学通报(2021年9期)2021-07-01
江苏农业科学(2016年8期)2017-02-15
广东海洋大学学报(2015年3期)2015-12-22
动物医学进展(2015年10期)2015-12-07
沈阳医学院学报(2014年4期)2014-12-27
淮阴工学院学报(2014年5期)2014-09-10
