
宽电压范围下阳极氧化制备TiO2纳米管阵列及其热稳定性
2010-11-09 10:43梁建鹤肖秀峰刘榕芳吴婷婷
无机化学学报 2010年1期
梁建鹤 肖秀峰 刘榕芳 俞 佳 吴婷婷
(福建师范大学化学与材料学院,福州 350007)
宽电压范围下阳极氧化制备TiO2纳米管阵列及其热稳定性
梁建鹤 肖秀峰 刘榕芳*俞 佳 吴婷婷
(福建师范大学化学与材料学院,福州 350007)
采用电化学阳极氧化技术,以含有NH4F和H2O的甘油溶液为电解液,在宽氧化电压范围(20~100 V)下于纯钛表面制备了结构高度有序的TiO2纳米管阵列。利用扫描电子显微镜(SEM)考察了阳极氧化工艺(氧化电压、NH4F浓度、环境温度、水分含量等因素)及退火处理对纳米管形貌的影响;采用X射线衍射分析(XRD)表征了不同氧化电压和退火前后TiO2纳米管阵列的物相;并从电流-时间曲线出发简要地分析了纳米管阵列的形成机理。结果表明,纳米管的内外径和管长随氧化电压的增大而增大;NH4F浓度和环境温度对纳米管形貌有一定的影响;水分含量的多寡决定了能否在高电压下自组装形成纳米管阵列;TiO2纳米管阵列具有良好的热稳定性,管状形貌可以保持到700℃;直接制备的TiO2纳米管阵列均为无定型结构,经450℃退火处理后,无定型的TiO2纳米管转变为锐钛矿相,而600℃退火处理后,部分锐钛矿相转变为金红石相。
阳极氧化;TiO2纳米管阵列;热稳定性
TiO2是一种典型的过渡金属氧化物,化学稳定性高、无毒、具有较高的催化活性和光电转化效率,因而在光催化、光电转化、光电催化、电致变色、储氢和感应器件等方面有着较为广泛的应用[1]。TiO2的纳米结构与其功能具有紧密的联系,因而制备具有特定形貌的TiO2纳米管成为材料领域最基础、最活跃的研究内容之一,也是实施TiO2纳米功能材料性质研究与技术开发的前提。已有文献[2-6]报道了采用阳极氧化技术在含氟电解液中于金属钛表面自组装形成排列规则的TiO2纳米管阵列,并可用于光电器件、光催化、染料敏化光电池(DSSC)和生物材料等领域。阳极氧化金属钛制备TiO2纳米管的方法具有工艺简单、纳米结构高度有序、管壁垂直于钛基底、管径和管长可控等优势,并且可在其管状结构中填充更小的有机、无机、金属和磁性粒子等物质组装成纳米复合材料,将进一步提高TiO2的光电、光催化以及其他方面的性能[7]。
由于TiO2纳米管阵列的结构是一端开口,一端闭合,呈试管状,内部存在空气且管径比较小,从而造成填充其他物质较为困难,因此制备大孔径的TiO2纳米管阵列有利于在纳米管内部填充其他物质,提高填充的效率。研究表明,TiO2纳米管阵列的孔径大小与阳极氧化的电压密切相关[2-3,6]。一般,随着阳极氧化的电压越高,TiO2纳米管阵列的孔径也随之增大,但氧化电压过高时,则无法形成自组装的纳米管阵列,取而代之的是无规则的多孔结构[2]。在HF或氟化物水溶液中,可制备纳米管的电压一般低于30 V,纳米管的孔径小于150 nm,管长较短。在有机溶剂中可制备纳米管的电压范围较宽,故可以获得一个较宽孔径范围的TiO2纳米管阵列,尤其在甘油体系(20~300 nm)[8]和二乙二醇体系(120~240 nm)[9]中。
对于甘油体系,Schmuki课题组[10-16]做了很多细致的研究,报道了以含0.5wt%NH4F的甘油为电解液可获得表面光滑的TiO2纳米管阵列,系统研究了甘油-NH4F和甘油-水-NH4F的混合溶液为电解液中工艺参数对纳米管形貌的影响,以及水分含量的影响,还分析了在上述体系中纳米管的形成过程。阴育新等[17-19]研究了甘油体系中不同的pH值和电导率对纳米管形成的影响,同时探讨了甘油和二甲亚砜(DMSO)的混合溶剂对阳极氧化过程中纳米管形成的影响,并且添加其他的阴离子到甘油溶液中,研究加入的阴离子对纳米管生长的影响。但均未对较高电压范围下的纳米管的形成行为进行研究。本文利用氧化电压和纳米管孔径的关系,以添加NH4F和H2O的甘油溶液为电解液选择较宽的电压范围阳极氧化纯钛制备出孔径分布更大的TiO2的纳米管阵列。同时研究了在拓展的电压范围下环境温度、NH4F浓度、水分含量和退火处理对纳米管表面形貌和长度的影响,并且讨论了不同制备条件和退火处理对纳米管晶相的影响。
1 实验部分
1.1 主要试剂与仪器
HF、HNO3、NH4F、甘油和丙酮等试剂均购自中国医药集团上海化学试剂公司,为分析纯试剂。二次蒸馏水为实验室自制。
扫描电子显微镜(SEM)(Philips XL30),透射电子显微镜 (TEM)(JEOL 1010),X射线衍射仪(XRD) (Philips X′Pert MPD),DF-101S型集热式恒温加热磁力搅拌器 (郑州长城科工贸有限公司),KQ218型超声清洗仪 (昆山市超声仪器有限公司),DH1715A-5型直流稳压电源 (北京大华无线电仪器厂),SX2-4-10型箱式电阻炉(上海实验电炉厂),DT9205N型数字万用表(深圳鸿昌滨江电仪器有限公司)。
1.2 TiO2纳米管阵列的制备及退火处理
将纯钛片(西北有色金属研究所,牌号TAl,技术条件GB/T3620.1-199A)裁剪成10 mm×100 mm,工作面积10 mm×10 mm,经1~6#金相砂纸打磨后,丙酮、二次水超声清洗和质量分数为4%HF-5 mol·L-1HNO3溶液浸蚀30 s后用蒸馏水洗净,干燥备用。
阳极氧化在敞开的常规二电极体系的电化学池中进行,阳极为预处理的钛片,大面积的铂片为对电极,电极间距保持在1 cm。整个实验过程在磁力搅拌和控制环境温度下进行。阳极氧化电源由DH1715A-5型直流稳压电源提供,以含0.125wt%~0.75wt%NH4F和0~10vol%H2O的甘油溶液为电解液,阳极氧化电压为20~100 V。阳极氧化24 h后,立即取出试样用二次水超声清洗并晾干。
为了研究TiO2纳米管阵列热稳定性和晶相变化,将阳极氧化过的钛片在箱式电阻炉中于450~700℃下退火处理,升温速率为5℃·min-1,在特定温度下保温2h后随炉自然冷却。
1.3 表征和测量
用 Philips XL30环境扫描电镜 (SEM)和JEOL1010透射电镜(TEM)观察试样的表面形貌和内部结构;用Philips X′Pert MPD X射线衍射仪(Cu Kα,管压40 kV,管流40 mA,步长0.02°,扫描速度4°·min-1)分析试样的物相,并且根据Scherrer公式[20]计算晶粒尺寸;用数字万用表记录阳极氧化过程中每个时刻的电流值,以此绘制电流-时间(j-t)曲线。
2 结果与讨论
2.1 TiO2纳米管阵列的形貌分析及主要影响因素
图1为纯钛在NH4F、H2O和甘油的混合溶液中不同氧化电压下阳极氧化制备的TiO2纳米管阵列的SEM照片。图2反映了氧化电压和纳米管的孔径和管长之间的关系。从图中可见,纳米管的管径随着氧化电压增加而增加,氧化电压为60V时纳米管阵列结构的规整程度最高。电压小于60 V时,内径和外径的增长速率分别约2.35和2.70 nm·V-1;电压大于60 V时,内径和外径的增长速率分别约5.24和8.87 nm·V-1。故当氧化电压大于60 V后,管壁厚度增加较快,同时管间距也较快的增大。当电压为90 V时,管内外径和管长分别增加到390、460 nm和约5 μm,从图1e的截面图看仍是排列紧密的纳米管阵列,是目前已报道的孔径最大的TiO2纳米管阵列。当电压为100 V时,管间距较大,形成的是分散的纳米管结构,同时可观测到分叉结构的纳米管(见图1f,分叉结构图中用黑线标出)。结果表明,阳极氧化电压是影响纳米管形貌和尺寸的重要因素,可通过控制阳极氧化电压来制备所需形貌和尺寸的纳米管阵列。

图1 甘油/水(90∶10,V/V)体系中不同氧化电压下TiO2纳米管阵列的SEM照片Fig.1 SEM top-view and cross-sectional(inset)images of TiO2nanotube arrays fabricated in a 0.5wt%NH4F and 10vol%H2O glycerol electrolyte for 24 h at 30℃with(a)20 V,(b)40 V,(c)60 V,(d)80 V,(e)90 V,and(f)100 V
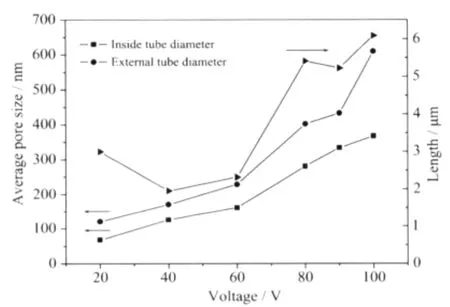
图2 TiO2纳米管阵列的管径和管长与氧化电压的关系Fig.2 Relation of anodic voltages and the morphology of TiO2nanotube arrays
图3为纯钛分别在60和100 V电压下阳极氧化制备的TiO2纳米管阵列的TEM照片。从图3(a)可见,TiO2纳米管呈中空结构且管壁较薄,其管外径约260 nm,平均的管壁厚度约12 nm,管外径与SEM观察到的基本匹配,外壁上有额外的环状物质突起。该环状突起物是由于在含较多水的电解液中阳极氧化会出现pH值周期突增的情况[8,15]。如果使用的电解液中含水量越低,则环状突起物越不明显,甚至在无水的条件下获得光滑管壁的纳米管[11]。从图3b中可观察到分叉结构的纳米管,证明了图1 (f)观察到分叉结构纳米管的事实。Mohapatra等[21]通过改变电解液的温度制备出分叉结构的纳米管,Valota等[16]利用程序升压的方法也制备出分叉结构的纳米管。而本研究表明,纳米管的分叉生长与氧化电压也有关。

图3 甘油/水(90∶10,V/V)体系中阳极氧化制备的TiO2纳米管阵列的TEM照片Fig.3 TEM images of TiO2nanotube arrays fabricated in a 0.5wt%NH4F and 10vol%H2O glycerol electrolyte for 24 h at 30℃with(a)60 V and (b)100 V
图4为纯钛在100 V电压下不同NH4F含量的混合溶液中阳极氧化制备的TiO2纳米管的SEM照片。从图中可见,在所考察的浓度范围内,100 V电压下制备的纳米管均为分散状,管间距较大,纳米管分叉生长现象明显。含0.75wt%NH4F的溶液中制备的纳米管分布极不均匀,纳米管以管簇的形式分散的分布在TiO2薄膜上,无纳米管覆盖的TiO2薄膜表面呈多孔形貌(见图4a,插图为多孔形貌的放大图)。含0.5wt%NH4F的溶液中制备的样品出现部分区域无管状结构覆盖 (见图1f)。含0.25wt% NH4F的溶液中得到的纳米管形貌与0.5wt%NH4F的相似,但无纳米管覆盖的区域进一步减小。可能是由于NH4F的浓度影响TiO2纳米管的化学腐蚀速率。随着NH4F浓度的提高,TiO2纳米管的化学腐蚀速率加快,当NH4F的浓度增大至0.75wt%时,纳米管大部分发生化学腐蚀溶解,表面变为不连续的孔状结构,而在较低NH4F浓度时,形成的纳米管比较密集,有管覆盖的区域较多。

图4 100 V电压下,不同NH4F浓度的溶液中制备的TiO2纳米管的SEM图Fig.4 SEM top-view and cross-sectional(inset)images of TiO2nanotubes fabricated for 24 h at 30℃with 100 V 10vol%H2O glycerol electrolytes

图5 60 V氧化电压,不同环境温度下制备的纳米管形貌的SEM照片Fig.5 SEM top view and cross sectional(inset)images of TiO2nanotube arrays fabricated at 60 V for 24 h in a 0.5wt%NH4F and 10vol%H2O glycerol electrolytes with(a)40℃,(b)50℃,and(c)60℃
图5为纯钛在不同环境温度下阳极氧化制备的TiO2纳米管阵列的SEM照片,其管径和管长随温度的变化如图6所示。从图中可见,随着环境温度的升高,纳米管的管内外径略微增加,而管长则是先增加再减小。一方面,环境温度升高将降低电解液的粘度,减小电解液的电阻,则加载在氧化膜层的电压提高,促进电场腐蚀,加快了纳米管的生长。因此,当环境温度从30℃升到40℃时,管长增加;另一方面,TiO2在含氟溶液中的化学腐蚀速率和环境温度有关,随着环境温度的升高,化学腐蚀的速率增加。当管口的腐蚀速率大于管的生长速率时,管长变短。故当温度高于40℃时,TiO2纳米管阵列的管长反而减小。
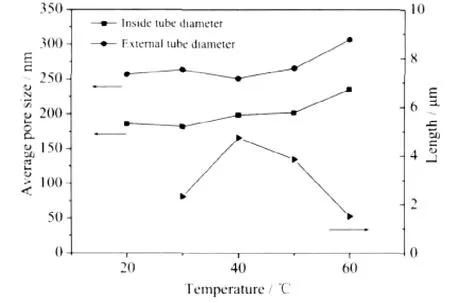
图6 60 V氧化电压下,不同温度对纳米管管径和管长的影响Fig.6 Influence of pore size and length of TiO2nanotubes fabricated at 60 V with different temperatures
图7为纯钛在不同水分含量的电解液中于100 V高电压下阳极氧化制备的样品的SEM照片。图7 (a)为不添加H2O的甘油溶液中,在100 V电压下阳极氧化制备的样品。可见,其没有得到规则的纳米管阵列,而是多孔的疏松结构,底部为水珠状凸起结构,从而造成该膜容易从基底上脱落,形成无支撑的透明薄膜。当电解液中添加5vol%H2O后,相同电压下氧化获得的是排列紧密的TiO2纳米管阵列(见图7(b)),其内径为350 nm,外径约410 nm,管长约2 μm。水分含量提高到10vol%时,相同电压下获得是分散的纳米管,管间距较大(见图1(f))。水分含量继续增加,则无法形成纳米管[19]。实验表明,适宜的水分含量有利于纯钛在高电压下阳极氧化获得排列紧密的纳米管阵列,水分含量过大过小都不利于形成纳米管。这可能是由于在纳米管形成的过程中需要从溶液体系中获得水分来支持纳米管的形成。水分的多寡会影响纳米管形成过程中的化学腐蚀速率,而纳米管阵列的形成是场致腐蚀和化学腐蚀相互协调匹配的结果。由于体系中水分缺乏,在蠕虫状的多孔层形成之后,无法在孔洞底部形成原位酸性。而纳米管形成过程中,孔洞底部原位酸性的存在是纳米管垂直于钛基底生长的必要条件[22]。由于高电压下孔洞底部的原位酸性条件在缺水分的条件下无法形成,无水甘油体系中孔洞生长缺乏径向的生长推动力,只能是无规律地向钛基地推进生长,最终形成水珠状的底部结构。并且水珠状突起的无规律性进一步证明了管状结构无法形成是由于缺乏径向生长的推动力造成的。同时,氟离子对形成的多孔层的溶解作用使得孔洞之间互相贯穿,最终形成类网状的多孔结构。因此,可以推测只有加入合适的水分含量才能促进孔洞底部原位酸性的形成,使得场致腐蚀和化学腐蚀处于一个合适的区间,促进纳米管阵列的形成。Prakasam等[23]也证明了在乙二醇体系中纳米管的形成也也存在着最佳的水分含量,仅有在合适的水分含量区间内,纳米管才能形成。

图7 100 V电压下,不同水分含量的溶液中所制备的纳米管的SEM照片Fig.7 SEM top view and cross sectional(inset)images of TiO2nanotube arrays fabricated at 100 V for 24 h in 0.5wt%NH4F glycerol electrolytes with the content of(a)0vol%H2O and(b)5vol%H2O
2.2 TiO2纳米管阵列的形成机理
一般认为在含有F-的偏酸性介质环境下,TiO2纳米管的形成过程主要发生了如下化学反应[2,3,22]:

阳极氧化过程中的电流-时间(j-t)曲线能够直接反映TiO2纳米管阵列的形成及变化过程。图8为纯钛在含0.5wt%NH4F和10vol%H2O的甘油溶液中于20、40、60、80、100 V电压下阳极氧化过程中的电流-时间(j-t)曲线。根据j-t曲线,整个氧化过程可大致分为3个阶段。第Ⅰ阶段,金属钛在含有F-的电解液中溶解,与水迅速反应,形成致密的氧化膜,导致电阻增大,电流快速降低。第Ⅱ阶段,由于氧化膜的形成,膜层承受的电场强度快速增大,在场致腐蚀和电解液对TiO2的化学溶解的共同作用下,氧化膜发生随机击穿溶解(反应式(3)),形成随机分布的表层孔核。随着氧化时间的增加,孔核发展成为蠕虫状的小孔,最后均匀分布在表面,导致该阶段阳极电流有所增大 (如图8,j-t曲线上电流呈轻微增大趋势)。由图8还可以观察到,随着氧化电压的增大,阳极电流的最小值依次降低,阳极电流出现增大的时间延长。这可能是由于电压越大,在第Ⅰ阶段形成的致密TiO2的厚度越大,导致阻挡层两侧的离子迁移越难,电流就越小[8]。TiO2的厚度越大,化学腐蚀减薄TiO2层所需的时间越长,电场腐蚀作用就越弱,导致第Ⅱ阶段电流增大时间延长。而当电压达到100 V时,几乎没有出现电流增大的趋势,这可能是由于电压太大,生成的致密TiO2的厚度很大,O2-透过薄膜在膜层表面结合Ti4+离子的速度,即形成TiO2膜的速率,与氟离子腐蚀TiO2膜的速率已达到接近稳定的状态 (即更早达到第Ⅲ阶段),致使电流无明显变化。第Ⅲ阶段,多孔膜的稳定生长,电流几乎全部由阻挡层两侧的离子迁移提供,并且由于甘油体系的粘度大,溶液中离子的迁移速率慢,从而形成一个小且稳定的电流值。由于微孔底部的电荷分布密度较孔壁大很多,使得孔底TiO2腐蚀速率较大,微孔不断地加深与加宽。此时,孔间的区域电荷密度增加且 TiO2的介电常数较小[22],促进了孔间氧化物的生长与溶解,于是在微孔之间形成了小空腔。纳米管微孔与空腔的协调生长便形成了纳米管阵列结构。当管底氧化层的生成速率和管口溶解速率平衡时,纳米管阵列的管长就不再增加。
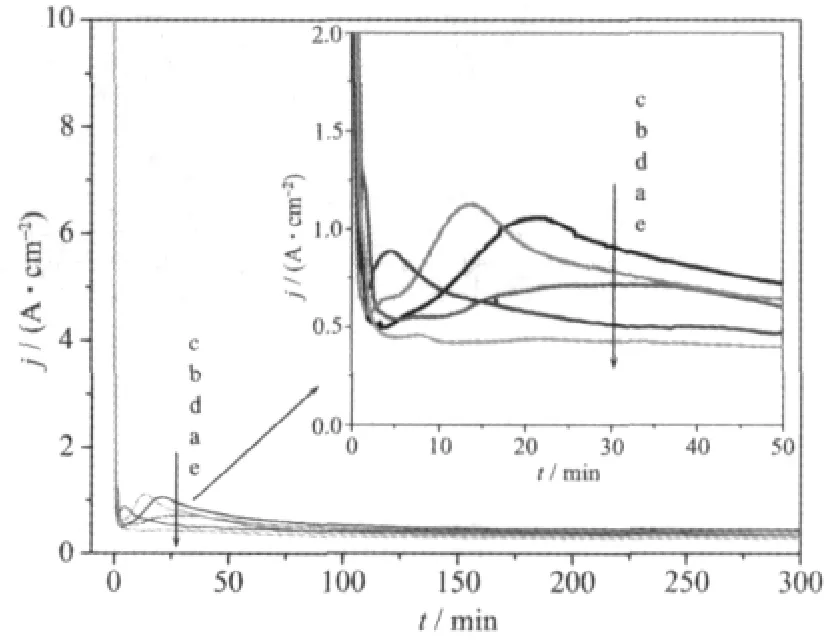
图8 甘油体系中阳极氧化过程的电流-时间(j-t)曲线Fig.8 j-t curves during anodic oxidation in glycerolbased electrolyte
在较高电压下氧化制备的样品容易制备得分叉结构的纳米管,如图1(f)和图3(b)所示。可能是由于在高电压下制备的纳米管的管径较大,并且底部的阻挡层较厚,在纳米管生长的过程中,可能在管底发生局部腐蚀不均匀的现象,持续一段时间后就会在纳米管底部形成若干个孔核。在电场辅助溶解效应和化学溶解的共同作用下,孔核生长成小孔,再进一步生长成分叉状的纳米管。
2.3 退火对TiO2纳米管阵列形貌和晶型的影响
本文还研究了退火处理对TiO2纳米管阵列形貌和晶型的影响。图9为纯钛在含0.5wt%NH4F和10vol%H2O的甘油溶液中60 V电压下阳极氧化24 h制备的样品经过不同温度退火处理后的SEM照片。图9(a)与图1(c)比较,可见经450℃退火后纳米管的形貌和尺度基本保持不变,管间的边界更清晰。从图9(b)和图9(c)可见,600和700℃退火后的样品的纳米管阵列结构基本保持完整,但表面一些比较脆弱的部分瓦解,轻微污染了纳米管表面。HF溶液中制备的TiO2纳米管阵列经过580℃退火后管状形貌已受影响,680℃已见明显的破坏[24]。Yang等[25]研究了无水甘油体系制备的TiO2纳米管阵列的热稳定性,结果表明TiO2纳米管阵列经700℃热处理后管状结构已无法分辨,并且管状结构的破坏是从底部开始的。与之比较,添加10vol%H2O的甘油溶液中制备的TiO2纳米管阵列显示了更好的热稳定性,管状阵列结构可以保持到700℃。

图9 60 V电压制备的TiO2纳米管阵列经不同温度退火后的SEM图Fig.9 SEM top view and cross sectional(inset)images of TiO2nanotube arrays fabricated at 60 V for 24 h in a 0.5wt%NH4F and 10vol%H2O glycerol electrolytes after annealed at(a)450℃,(b)600℃,and(c)700℃
图10为不同氧化条件和不同温度退火处理后TiO2纳米管阵列的XRD图。图10中曲线a和b仅出现钛的衍射峰,说明阳极氧化直接制备的TiO2纳米管阵列是无定型的,氧化电压对纳米管晶相影响很小。无定型的TiO2纳米管阵列经450℃退火处理后,出现了尖锐的锐钛矿相TiO2(PDF No.73-1764)的衍射峰(图8(c)),晶粒尺寸为33.1 nm;退火温度升到600℃时,锐钛矿型TiO2的峰减弱,并出现了较弱的金红石相TiO2(PDF No.65-0190)的衍射峰(图8d)。样品中金红石相的质量分数可按下式估算[26]:
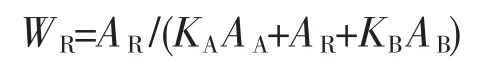
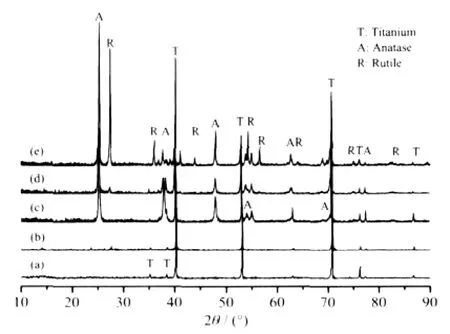
图10 不同氧化条件下和退火后的氧化钛纳米管阵列的XRD图Fig.10 XRD patterns of TiO2nanotube arrays fabricated in a 0.5wt%NH4F and 10vol%H2O glycerol electrolyte at different anodizing conditions of (a)60 V with 30℃,(b)100 V with 30℃,and 60 V with 30℃after annealed at(c)450℃, (d)600℃,and(e)700℃
其中,AA、AR和AB分别为锐钛矿(101)、金红石(110)和板钛矿(121)晶面衍射峰的强度。KA和KB为常数,分别为0.886和2.721。此时,金红石相占15.3%,锐钛矿相占84.7%。当退火温度上升到700℃后,金红石相TiO2的衍射峰显著增强,金红石相占48.1%,锐钛矿相占51.9%。随着退火温度从450℃升到600℃时,锐钛矿相的微晶尺寸基本不变,约33.9 nm,此时金红石相的晶粒尺寸为12.5 nm。当退火温度升高到700℃时,锐钛矿相和金红石相的晶粒尺寸都明显变大,各增大到68.0和43.4 nm,晶粒尺寸的变化规律与文献[24]相符。 肖秀峰等[27]认为TiO2纳米管阵列退火后管状形貌的崩溃与TiO2退火过程中的晶型转变和钛基底的氧化有关。TiO2纳米管阵列在锐钛矿相与金红石相转变的过程中,会出现晶体的崩溃和重组,从而导致了TiO2纳米管阵列形貌的崩溃。故TiO2晶型转变温度是管状形貌能在高温下保持的关键因素。HF溶液中制备的TiO2纳米管阵列在430℃退火后就出现金红石相[24,28],而本体系制备的TiO2纳米管阵列金红石相出现的退火温度为600℃,这可能是导致了其管状结构能保持到700℃的重要原因。
3 结 论
(1)通过调节氧化电压可在含0.5wt%NH4F和10vol%H2O的甘油溶液中阳极氧化制备得孔径在100~460 nm之间连续可调的TiO2纳米管阵列。工艺研究表明,在相同的氧化时间内,纳米管的管径和管长随氧化电压的增大而增大;氧化电压为100 V时,纳米管以分散的纳米管形式分散在钛基底上,同时纳米管存在明显的分叉结构;提高环境温度,可使制得的纳米管的孔径变大,但管长先增大后明显减小;适宜的水分含量是在高电压下制备紧密排列的TiO2纳米管阵列的关键。
(2)TiO2纳米管阵列在空气中退火处理会发生一系列的晶相转变。450℃退火后无定型转变为锐钛矿相,600℃退火后出现金红石相,700℃退火后金红石相比例升高,此时纳米管阵列结构仍然保持完整,显示了较高的热稳定性。
[1]Chen X B,Mao S S.Chem.Rev.,2007,107(7):2891-2959
[2]Mor G K,Varghese O K,Paulose M,et al.Sol.Energy Mater. Sol.Cells,2006,90(14):2011-2075
[3]Macak J M,Tsuchiya H,Ghicov A,et al.Curr.Opin.Solid State Mater.Sci.,2007,11(1/2):3-18
[4]SUN Lan(孙 岚),LI Jing(李 静),ZHUANG Hui-Fang(庄惠芳),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao), 2007,23(11):1841-1850
[5]Grimes C A.J.Mater.Chem.,2007,17(15):1451-1457
[6]Ghicov A,Schmuki P.Chem.Commun.,2009,36:2791-2808
[7]Raja K S,Misra M,Paramguru K.Mater.Lett.,2005,59(17): 2137-2141
[8]Macak J M,Hildebrand H,Marten-Jahns U,et al.J.Electroanal.Chem.,2008,621(2):254-266
[9]Yoriya S,Mor G K,Sharma S,et al.J.Mater.Chem.,2008, 18(28):3332-3336
[10]Macak J M,Aldabergerova S,Ghicov A,et al.Phys.Stat. Sol.(a),2006,203(10):R67-R69
[11]Macak J M,Luciano Taveira H T,Aldabergerova S,et al. Angew.Chem.Int.Ed.,2005,44(45):7463-7465
[12]Berger S,Macak J M,Kunze J,et al.Electrochem.Solid-State Lett.,2008,11(7):C37-C40
[13]Macak J M,Albu S,Kim D H,et al.Electrochem.Solid-State Lett.,2007,10(7):K28-K31
[14]Macak J M,Schmuki P.Electrochim.Acta,2006,52(3):1258 -1264
[15]Valota A,LeClere D J,Skeldon P,et al.Electrochim.Acta, 2009,54(18):4321-4327
[16]Valota A,LeClere D J,Hashimoto T,et al.Nanotechnology, 2008,19:355701
[17]Yin Y X,Jin Z G,Hou F,et al.J.Am.Ceram.Soc.,2007, 90(8):2384-2389
[18]YIN Yu-Xin(阴育新),JIN Zheng-Guo(靳正国),TAN Xin(谭欣),et al.Acta Phys.-Chim.Sin.(Wuli Huaxue Xuebao), 2008,24(11):2133-2138
[19]YIN Yu-Xin(阴育新),JIN Zheng-Guo(靳正国),HOU Feng (侯 峰).Acta Phys.-Chim.Sin.(Wuli Huaxue Xuebao), 2007,23(11):1797-1802
[20]Scherrer P.Nachr.Ges.Wiss.Göttingen,1918,26:98-100
[21]Mohapatra S K,Misra M,Mahajan V K,et al.Mater.Lett., 2008,62(12/13):1772-1774
[22]Tao J L,Zhao J L,Tang C C,et al.New J.Chem.,2008,32: 2164-2168
[23]Prakasam H E,Shankar K,Paulose M,et al.J.Phys.Chem. C,2007,111(20):7235-7241
[24]Varghese O K,Gong D,Paulose M,et al.J.Mater.Res., 2003,18(1):156-165
[25]Yang Y,Wang X H,Li L T.J.Am.Ceram.Soc.,2008,91 (2):632-635
[26]Zhang H,Banfield J F.J.Phys.Chem.B,2000,104(31): 3481-3487
[27]XIAO Xiu-Feng(肖秀峰),LI Ming-Ou(李明欧),LIU Rong-Fang(刘榕芳),et al.Rare Metal Mater.Eng.(Xiyou Jinshu Yu Gongchen),2008,37(7):1245-1249
[28]Ge R,Fu W,Yang H,et al.Mater.Lett.,2008,62(18/19): 2688-2691
Fabrication and Thermal Stability of TiO2Nanotube Arrays by Anodic Oxidation at Wide Range of Voltage
LIANG Jian-He XIAO Xiu-Feng LIU Rong-Fang*YU Jia WU Ting-Ting
(College of Chemistry and Materials Science,Fujian Normal University,Fuzhou350007)
Anodic oxidation was adopted to prepare TiO2nanotube arrays,which were prepared isobarically by the anodization of pure Ti in glycerol-based electrolyte containing NH4F and H2O at wide range of anodic voltage.The effect of anodic voltages,fluorine concentration,environmental temperature,the content of H2O and thermal annealing treatment on the morphology of TiO2nanotube was studied.The titania nanotube arrays were characterized with SEM and XRD.Then,the formation mechanism of nanotube arrays based on the current-time curves was suggested.The results showed that inside and external diameter increased with the increase of voltage;fluorine concentration and environment temperature could effected the morphology of TiO2nanotubes;the content of H2O effected the grown nanotube at high voltage.As-prepared TiO2nanotube arrays has good thermal stability.Theirs tubular morphology can maintain higher than 700℃.After annealing at 450℃,the amorphous nanotubes changed into anatase type.After annealing at 600℃,part of anatase type changed into rutile.
anodic oxidation;TiO2nanotubes arrays;thermal stability
O614.41+1;O646.6
A
1001-4861(2010)01-0112-08
2009-07-20。收修改稿日期:2009-09-07。
国家自然科学基金(No.3060149;30970887),卫生部科学研究基金(No.WKJ2008-02-037)资助。*
。E-mail:rfliu@vip.sina.com,Tel:+86591-83465190
梁建鹤,男,25岁,硕士研究生;研究方向:纳米生物陶瓷材料。
猜你喜欢
四川地质学报(2022年2期)2022-07-08
矿产勘查(2020年8期)2020-12-25
原子与分子物理学报(2020年5期)2020-03-17
——以金红石为例
中国金属通报(2020年23期)2020-03-15
节水灌溉(2019年11期)2019-11-28
无机盐工业(2017年6期)2017-03-11
无机盐工业(2016年6期)2016-03-15
无机盐工业(2016年2期)2016-03-15
安徽大学学报(自然科学版)(2015年1期)2015-12-05
工业设计(2015年12期)2015-10-21
