
DNA甲基化异常与肿瘤耐药
2014-05-25 00:32司鑫鑫孙玉洁
遗传 2014年5期
司鑫鑫, 孙玉洁
江苏省人类功能基因组学重点实验室, 南京医科大学基础医学院细胞生物学系, 南京210029
DNA甲基化异常与肿瘤耐药
司鑫鑫, 孙玉洁
江苏省人类功能基因组学重点实验室, 南京医科大学基础医学院细胞生物学系, 南京210029
肿瘤耐药是导致肿瘤化疗失败的主要原因, 其产生机制复杂多样, 是多种因素共同作用的结果。近年来,表观遗传改变在肿瘤耐药中的作用日益受到关注。DNA甲基化是一种重要的表观遗传修饰, 在调节基因表达和维持基因组稳定性中扮演着重要角色。原发性或获得性耐药的肿瘤细胞大多伴随 DNA异常甲基化, 越来越多的证据显示, DNA甲基化异常是肿瘤细胞耐药表型产生的重要机制。文章就DNA甲基化异常与肿瘤细胞耐药的关系及相关作用机制进行了综述。
肿瘤耐药; DNA甲基化; 基因组稳定性; 基因表达
恶性肿瘤是危害人类健康的重大疾病, 化疗是治疗恶性肿瘤的重要手段之一。尽管随着新药的问世和医疗水平的提高, 肿瘤治疗水平得到极大提高,但肿瘤耐药依然是临床肿瘤治疗的主要障碍之一,其机制复杂多样, 是多种因素共同参与的结果[1,2],如细胞抗凋亡能力增强、DNA损伤修复能力异常、ATP依赖性药物转运泵表达上调、药物代谢改变等,这其中涉及一系列基因表达调控的异常。越来越多的证据显示, DNA甲基化异常是肿瘤细胞耐药表型产生的重要机制。认识DNA甲基化改变在肿瘤耐药性形成过程中的作用, 对于更好地认识肿瘤耐药的产生机制, 进而有效预防和逆转耐药表型具有重要的意义。
1 特定基因的甲基化异常与肿瘤耐药
化疗药物通过引起DNA损伤、抑制增殖、促进细胞凋亡等多种途径杀死肿瘤细胞, 如:铂类药物可引起DNA链间或链内交联, 造成DNA断裂, 抑制DNA和RNA合成, 抑制细胞有丝分裂; 喜树碱类化疗药物可抑制拓扑异构酶Ⅰ, 阻止 DNA解旋,造成DNA损伤, 诱导细胞凋亡; 植物类药物如长春新碱、紫杉醇等, 可与微管结合影响其稳定性, 使有丝分裂停滞。肿瘤细胞内上述相关通路中的基因启动子甲基化水平异常所致表达改变均可能影响肿瘤细胞对化疗药物的敏感性。
1.1 DNA损伤修复基因的异常甲基化与肿瘤耐药
烷化剂、铂类等多种化疗药物能破坏DNA结构引起DNA损伤, 带有DNA损伤的细胞必须在细胞周期中暂停, 以便得到及时修复。DNA损伤修复是恢复 DNA正常序列结构和维持遗传信息相对稳定的细胞反应, 包括错配修复、切除修复、双链断裂修复等。DNA损伤修复能力的改变是多种化疗药物耐药的重要分子基础。
DNA错配修复系统在肿瘤细胞中常表现为功能异常。错配修复系统的失活, 除了在多种肿瘤的发生过程中发挥重要作用, 也是导致恶性肿瘤耐药的一个重要因素。错配修复系统失活使细胞监测DNA损伤的能力下降, 因而不能激活凋亡反应; 基因突变率升高, 基因组不稳定性增加, 直接或间接地促进肿瘤耐药表型的形成[3,4]。DNA甲基化升高可以导致错配修复基因失活, 加强肿瘤细胞耐药性。例如:在顺铂耐药的卵巢癌细胞中, 编码DNA错配修复酶的hMLH1基因启动子的甲基化水平升高, 基因表达水平下降; 用甲基转移酶抑制剂处理该细胞,可恢复hMLH1表达, 并能部分恢复耐药细胞对顺铂的敏感性[5,6]。比较临床卵巢癌患者在化疗前和复发时的血浆hMLH1启动子甲基化水平, 发现25%的患者在化疗前无hMLH1甲基化而复发时hMLH1发生甲基化并伴有微卫星不稳定性[7]。
乳腺癌易感基因 BRCA1被认为是基因组的看护者, 在DNA损伤修复、细胞周期调控等过程中发挥重要作用[8]。当细胞内发生DNA双链断裂, H2AX快速磷酸化, 将BRCA1募集到DNA损伤位点, 与Rad51、BRCA2、Rad50/Mre11/p95等相互作用, 对损伤 DNA进行修复[9,10]。有研究报道, BRCA1的DNA损伤修复功能参与了肿瘤耐药。在MCF7的顺铂耐药细胞中BRCA1高表达, DNA修复能力明显增强; 用siRNA下调耐药细胞中BRCA1表达水平, 顺铂处理所产生的DNA损伤不能被及时修复, 凋亡增多, 细胞对顺铂的耐药性下降[11]。临床研究也发现,乳腺癌和卵巢癌患者的 BRCA1表达水平可作为化疗药物的选择依据, BRCA1功能缺失的肿瘤细胞对DNA靶向的化疗药物及纺锤体毒性药物更敏感[12]。启动子区甲基化异常是造成 BRCA1表达改变的主要原因之一[13]。BRCA1基因启动子区的甲基化水平和卵巢癌患者对铂类化疗的敏感性呈正相关[14]。
1.2 凋亡相关基因的异常甲基化与肿瘤耐药
多数化疗药物可通过诱导细胞凋亡来杀死肿瘤细胞, 反之, 肿瘤细胞也可通过逃逸或拮抗凋亡获得生存[15]。凋亡酶激活因子-1(APAF1)是一种促凋亡基因, 在线粒体参与的凋亡途径中具有重要作用。APAF1阴性的黑色素瘤细胞对各种化疗药物均有不同程度的耐受, 用DNA甲基转移酶抑制剂处理, 可恢复其表达, 肿瘤细胞对化疗的敏感性也随之升高[16,17]。CASP8也是一种促凋亡基因, 是死亡受体介导的凋亡途径中的关键启动子。研究发现, 在多种肿瘤细胞中 CASP8基因的启动子均被甲基化, DNA甲基转移酶抑制剂处理可逆转 CASP8的高甲基化, 提高肿瘤细胞对多种化疗药物如阿霉素、依托泊苷、顺铂等的敏感性[18]。Chaopatchayakul等[19]检测了化疗敏感和耐受的宫颈癌患者样本中一系列凋亡基因的甲基化水平, 发现化疗耐受组患者的死亡相关蛋白激酶基因(DAPK)和肿瘤坏死因子受体超家族成员(FAS)的甲基化水平更高。DAPK和 FAS基因的高甲基化导致其表达沉默, 使肿瘤细胞可以逃逸化疗药物引起的凋亡反应从而获得生存。肿瘤细胞内凋亡信号通路上相关基因的甲基化异常可引起凋亡反应能力的改变, 治疗前及治疗过程中监测这些基因的甲基化水平, 可为优化肿瘤患者化疗方案及预后评估提供重要依据。
1.3 药物转运及药物代谢基因的异常甲基化与肿瘤耐药
ATP结合盒(ATP-binding cassette, ABC)转运蛋白是ATP依赖性药物外排泵, 利用ATP作为能源跨膜转运许多物质, 如离子、氨基酸、蛋白质、糖类、磷脂和药物等, 其表达或功能异常是肿瘤细胞产生多药耐药的重要机制。目前已知人类基因组共包含48个具有转录活性的ABC转运子基因, 其中与多药耐药相关的主要有MDR1(ABCB1)、MRPs(ABCC家族)、BCRP(ABCG2)等[20]。药物转运基因启动子的低甲基化可以促进这些基因的表达, 增强细胞泵出药物的能力, 降低肿瘤细胞内的药物浓度, 削弱药物的细胞毒性[20]。临床资料显示, 膀胱癌病人化疗后MDR1的表达比化疗前升高3.5~5.7倍, 89%的复发病人MDR1高表达, 且MDR1的表达水平与基因启动子甲基化水平呈显著负相关[21]。Kusaba等[22]用长春新碱和阿霉素处理鳞状细胞癌KB3-1诱导得到长春新碱耐药细胞 KB/VJ300和阿霉素耐药细胞KB-C1, 发现与敏感细胞相比, 两株耐药细胞中MDR1基因启动子甲基化水平下降, 其表达显著升高。ABCG2是另一个与耐药有关的转运蛋白。ABCG2高甲基化的肿瘤细胞对其底物化疗药物如米托蒽醌、拓扑替康等敏感, 而ABCG2低甲基化的肿瘤细胞对这些化疗药耐受[23]。Bram等[24]发现用柳氮磺吡啶和拓扑替康处理白血病细胞和卵巢癌细胞12~24 h可诱导ABCG2基因启动子的完全去甲基化,使ABCG2转录升高235倍从而获得ABCG2依赖性的耐药表型, 这表明化疗药处理后 ABCG2的表观激活是肿瘤细胞耐药性产生的早期事件。其他一些研究也都发现类似结果[25], 即化疗药物处理可引起肿瘤细胞内 ABC家族基因的启动子甲基化水平下降从而表达升高, 获得更强的药物泵出能力, 产生化疗耐药。
药物进入体内, 会经过一系列复杂的过程, 包括药物的吸收、分布、代谢、排泄等。多数药物经过代谢后药理作用减弱或消失, 少数药物经过代谢变为活性形式发挥治疗作用。药物代谢酶(Cytochrome P450 proteins, CYP)的表达紊乱或活性变化, 是引起肿瘤耐药的重要因素。长春碱类化疗药物经 CYP3A4代谢后为无毒性形式, 在大肠癌细胞 LS174T中诱导 CYP3A4表达后, 可明显增强细胞对长春花碱的耐受性[26]。另外, CYP3A可以通过减弱紫杉醇的药理活性, 使大肠癌细胞耐受[27]。CYP基因在不同情况下(如肿瘤、吸烟等)甲基化水平会发生改变[28,29], 但耐药肿瘤细胞中CYP基因的异常表达是否由启动子甲基化改变所致, 则需要进一步的研究证实。
1.4 肿瘤干细胞标志基因的异常甲基化与肿瘤耐药
肿瘤干细胞学说认为肿瘤组织中存在一群比例很小但具有自我更新能力和无限增殖潜能的细胞,即肿瘤干细胞, 它们不仅是肿瘤发生的始动细胞,而且可能是导致肿瘤耐药的主要原因[30,31]。已知干性因子OCT4、SOX2等在肿瘤干细胞形成和干性维持过程中发挥重要作用[32~35], 且这些基因的表达都受甲基化调控[36,37], 提示干细胞标志基因的甲基化水平改变可能会影响肿瘤细胞对化疗药物的敏感性。Wang等[38]发现耐药的肝癌组织中OCT4转录水平明显升高; 在敏感细胞内过表达或在耐药细胞内干扰 OCT4表达, 可相应增强或降低肿瘤细胞对化疗药物的耐药性。进一步实验发现, 耐药肿瘤细胞内OCT4启动子甲基化水平下降导致了OCT4转录水平的升高。这些结果提示, DNA甲基化水平异常可能通过影响肿瘤干细胞形成或维持进而改变化疗敏感性。
总之, 细胞的多种生物学机制是否正常有效地发挥作用, 决定了细胞对外界环境包括药物的反应性。肿瘤细胞在其形成过程以及接受药物处理过程中发生的表观遗传学改变, 在很大程度上赋予部分肿瘤细胞特定的生存优势, 形成耐药表型。与肿瘤耐药相关的一些异常甲基化改变见表1。
2 全基因组甲基化水平异常与肿瘤耐药
耐药的肿瘤细胞内除了发生某些特定基因的甲基化水平异常, 同时还会发生全基因组甲基化水平的异常。Nyce等[42]选用多种化疗药物处理肺癌细胞HTB-54和横纹肌肉瘤细胞 CCI-136, 发现依托泊苷、阿霉素、长春新碱、顺铂、5-氟尿嘧啶、甲氨喋呤等可使HTB-54和CCI-136的全基因组甲基化水平明显升高。Kastl等[43]用多西紫杉醇诱导MDAMB-231和 MCF7细胞得到两株多西紫杉醇耐药细胞, 检测发现 MCF7耐药细胞中全基因组甲基化水平升高, 而 MDA-MB-231耐药细胞中略下降。Segura-Pacheco等[44]在 MCF7的阿霉素耐药细胞中也检测到全基因组甲基化水平升高, 降低全基因组甲基化水平可部分恢复耐药细胞对阿霉素的敏感性。本实验室也观察到类似现象, 与敏感细胞相比, MDA-MB-231的紫杉醇耐药细胞中全基因组甲基化水平下降; 而在 MCF7的紫杉醇耐药细胞中全基因组甲基化水平升高[45]。
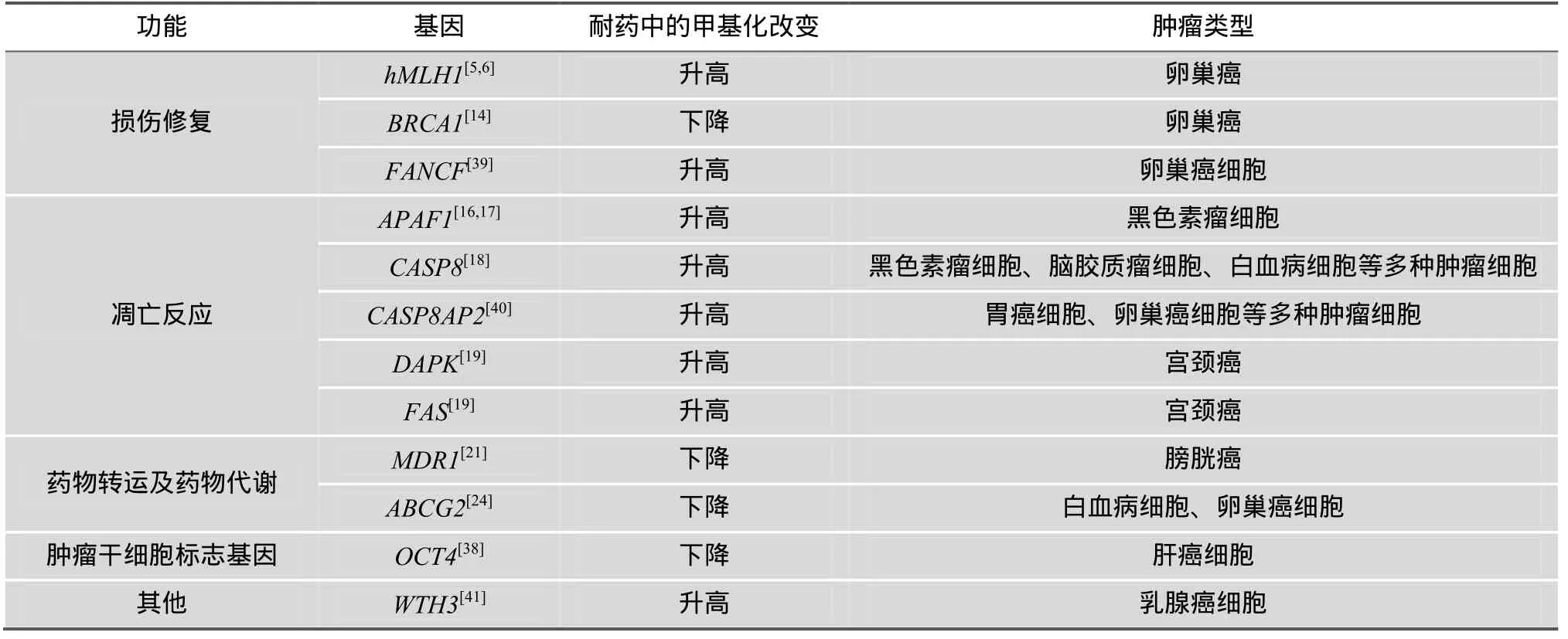
表1 与肿瘤耐药相关的异常甲基化
化疗药物引起基因组 DNA甲基化水平改变的机制尚不清楚。Nyce等[42]认为化疗药物会引起全基因组甲基化水平升高可能是因为这些药物能引起复制叉停滞, DNA甲基转移酶(DNA methyltransferases, DNMTs)与新合成的DNA作用时间延长, 更多的胞嘧啶发生甲基化, 其研究结果也证实了上述推测。
DNA甲基转移酶的表达改变也能直接影响全基因组甲基化水平。DNA甲基转移酶的表达受多种转录因子调控, 如雌激素受体(Estrogen receptor alpha, ERα)。Cui等[46]发现, ERα可增强DNMT3b基因的转录活性。用 10-8M的雌激素处理子宫内膜癌细胞可上调DNMT3b的表达, 细胞的DNA从头甲基化能力增强; 用 ERα抑制剂预处理, 可抑制雌激素引起的 DNMT3b过表达, 表明雌激素激活DNMT3b是ERα依赖性的。本实验室研究结果发现, ERα可增强 DNMT1基因启动子的活性[45]。ERα在很多耐药的肿瘤细胞中表达异常, ERα表达或活性的改变会引起其靶基因DNMT1和DNMT3b的表达异常, 进而影响全基因组甲基化水平。由此可以推测, 任意一种影响ERα的表达或活性的药物均可能影响细胞内全基因组甲基化水平, 进而影响细胞对药物的反应性。
全基因组甲基化水平的升高或下降都会影响肿瘤细胞对化疗药物的敏感性。全基因组异常高甲基化对肿瘤耐药的作用机制目前还没有很好的阐述,可能通过沉默特定基因进而干扰DNA损伤修复、细胞凋亡和细胞周期等关键信号通路促进肿瘤耐药。全基因组异常低甲基化可造成基因组不稳定[47~49],促进耐药亚群细胞的产生, 作用机制尚不很清楚,可能通过以下两方面:(1)激活反转座元件:分散在真核生物基因组中的大量逆转座子是引起基因组不稳定的重要因素, 可引起基因序列缺失、扩增、易位等[50~52]。LINE-1是一种自主性逆转座子, 在人类基因组中广泛分布, 能编码ORF1和ORF2两种蛋白,其中ORF2具有核酸内切酶活性, 可以导致DNA双链断裂以及基因组不稳定。在大多数情况下, LINE-1启动子因高度甲基化而处于沉默状态, 但在某些特殊情况下 LINE-1启动子可发生去甲基化而被激活; (2)中心粒周围异染色质去凝集, 形成可以重组的染色质构象[53]:异染色质中的组蛋白去乙酰化和DNA高度甲基化使其处于高度压缩致密状态, 当基因组DNA去甲基化, 异染色质区结构松散, 基因组不稳定性升高[54,55]。目前认为全基因组异常低甲基化会促进基因组不稳定, 但有趣的是, 本课题组在全基因组甲基化升高的MCF7耐药细胞中也发现了拷贝数变异显著增加, 显示基因组不稳定性升高。显然, 全基因组甲基化水平应处在一个稳态范围内,过高或过低都将削弱基因组稳定性。
基因组不稳定性是一种以遗传改变频率升高为特点的细胞状态, 包括突变、重复或缺失、易位、异倍体等[56]。在许多肿瘤中, 如乳腺癌、大肠癌、肺癌, 基因组不稳定性与原发性耐药、获得性耐药以及肿瘤的不良预后相关[57~59]。肿瘤细胞的基因组不稳定有利于细胞在面对外界不利的生存压力时产生适应性改变从而存活, 并且这种改变是可以遗传的。恶性细胞的演化本质上都符合达尔文进化规律,基因组不稳定性和肿瘤所处微环境的选择压力, 使肿瘤细胞处在不断的进化演变中[60]。基因组不稳定所产生的肿瘤异质性是肿瘤进化所需的原材料, 而选择压力则是加速这一过程的推动力。化疗药物的存在对肿瘤细胞群体是一种强烈的选择压力, 这种选择压力会加快进化速度, 将本来存在的适应性克隆筛选富集出来(原发性耐药), 或者诱导产生新的适应性改变(获得性耐药)。
全基因组 DNA甲基化异常与特定基因甲基化异常在肿瘤耐药性形成过程中并不是相互排斥、相互独立, 而是相辅相成、相互影响。重复序列LINE-1约占人类基因组的 20%[61], 其甲基化水平可在相当程度上代表全基因组甲基化水平[62,63]。LINE-1可位于基因间也可位于基因内, 据统计, 有超过2000个活性LINE-1位于基因内[64]。位于基因内部的LINE-1被认为是一种顺式调控元件, 其调节基因表达的主要机制之一是LINE-1自身表观修饰的改变[61]。位于基因启动子区的LINE-1甲基化水平的变化, 可影响基因启动子活性进而调节其表达。特定基因的甲基化异常是整体基因组甲基化水平异常的部分体现,全基因组甲基化异常也会影响特定基因的甲基化水平。
肿瘤细胞中的DNA甲基化模式发生改变, 这些改变不仅在肿瘤的发生发展过程中发挥重要作用,也是导致肿瘤耐药的重要因素。如, 错配修复基因hMLH1的异常高甲基化被认为在微卫星不稳定性结直肠癌的形成过程中发挥重要作用, 是其癌变过程中的早期事件[65]; hMLH1异常甲基化也是导致肿瘤耐药的重要因素, 错配修复系统失活使肿瘤细胞监测DNA损伤的能力下降, 不能激活凋亡反应, 对化疗药物耐受性增强[7,66]。促凋亡基因APAF-1异常高甲基化所致细胞凋亡能力下降, 是促进肿瘤形成和肿瘤耐药的重要分子机制[17]。然而, 肿瘤发生过程中产生的 DNA甲基化改变与耐药过程中发生的DNA甲基化改变不可能完全相同, 是由于这两种过程所涉及的细胞生物学改变和分子机制不尽相同,导致其改变的表观遗传修饰必有差异。例如, BRCA1是一个具有多重功能的蛋白, 在DNA损伤修复、转录调控中的功能最为显著。BRCA1基因启动子的高甲基化导致其表达下降, 与乳腺癌和卵巢癌的发生密切相关[13,67], 而BRCA1基因启动子的低甲基化改变与肿瘤耐药密切相关, BRCA1甲基化程度越低,表达越高, 细胞损伤修复能力越强, 对铂类化疗药物的耐受性越好[14]。因此鉴定肿瘤发生发展过程及肿瘤耐药过程中的DNA异常甲基化改变, 具有重要的理论意义和临床应用潜力。
3 对克服肿瘤耐药的提示
DNA甲基化是一种可逆的表观修饰, 认识和探讨DNA甲基化与肿瘤耐药的关系及机制, 为临床上控制肿瘤耐药提供了新的可能性。一种可能是利用一些靶向 DNA甲基转移酶的小分子化合物调节甲基转移酶活性, 改变基因启动子的甲基化状态从而逆转耐药。例如:地西他滨是一种胞嘧啶脱氧核糖类似物, 能整合到DNA分子中, 与DNA甲基转移酶形成加合物抑制其活性。除了在临床骨髓增生异常综合征和白血病的治疗中取得了很好的疗效[68,69],地西他滨还可以降低 hMLH1基因启动子甲基化水平, 恢复细胞错配修复功能, 从而逆转卵巢癌细胞A2780/cp70对DDP的耐药[5]。美国食品和药品管理局已于2007年批准地西他滨用于临床试验, 同期批准使用的还有维达扎, 一种胞嘧啶核糖类似物[70]。另一种控制肿瘤耐药的可能途径是抑制或降低DNA甲基转移酶的表达水平。已知DNA甲基转移酶的表达受多种转录因子调控, 如雌激素受体 ERα可调节DNMT1和DNMT3b的转录水平[45,46], 因此可利用ERα拮抗剂间接调控基因组DNA 甲基化水平, 如能特异性下调ERα表达的氟维司群[71]。关于ERα及其拮抗剂氟维司群在乳腺癌耐药中的作用已有一些研究, ERα与基因组甲基化改变及耐药的关系和机制值得进一步探讨。
通过靶向 DNA甲基转移酶逆转肿瘤耐药因特异性差而受到很大局限性, 在用于临床治疗时会产生较大的毒副作用。因此寻找特异性改变DNA甲基化的方法显得尤为重要。最新研究发现, 某些长链非编码RNA能与DNMT1蛋白相互作用, 改变靶基因的DNA甲基化水平[72]。CEBPA基因可以转录一种不含 polyA 的额外编码的 RNA(extra-coding CEBPA, ecCEBPA), 这种RNA在细胞核内富集, 不被翻译。当 DNMT1靠近 CEBPA基因启动子时, ecCEBPA能与DNMT1蛋白相互作用, 阻止DNMT1与启动子的结合, 防止CEBPA基因被甲基化, 上调其转录水平。这种相互作用不只局限于CEBPA基因,在细胞内存在很多这种与 DNMT1相互作用的不含polyA的 RNA, 能调节特定基因的甲基化水平。暂不考虑ecRNA如何产生或用于临床治疗的可操作性如何, 它的出现意味着特异性改变靶基因的甲基化水平成为可能。
4 结语与展望
DNA甲基化异常在肿瘤耐药性形成过程中发挥着重要作用。鉴于DNA甲基化可能影响不同的基因, 产生完全相反的效应, 因此, 利用 DNA甲基化机制逆转肿瘤耐药面临着巨大挑战, 即确定具有生物标志物性质的甲基化模式。这意味着必须确定哪些或哪一组基因, 或什么程度的基因组甲基化改变是特定肿瘤细胞耐药的驱动力, 哪些甲基化变化仅仅是伴随出现的现象, 这将帮助确定哪些患者适合表观遗传治疗, 这一目标需要遗传学研究和临床医生的联手努力方可达成。
[1] Raguz S, Yague E. Resistance to chemotherapy: new treatments and novel insights into an old problem. Br J Cancer, 2008, 99(3): 387–391.
[2] Mellor HR, Callaghan R. Resistance to chemotherapy in cancer: a complex and integrated cellular response. Pharmacology, 2008, 81(4): 275–300.
[3] Aebi S, Kurdi-Haidar B, Gordon R, Cenni B, Zheng H, Fink D, Christen RD, Boland CR, Koi M, Fishel R, Howell SB. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res, 1996, 56(13): 3087–3090.
[4] Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res, 1998, 4(1): 1–6.
[5] Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res, 2000, 60(21): 6039–6044.
[6] Strathdee G, MacKean MJ, Illand M, Brown R. A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer. Oncogene, 1999, 18(14): 2335–2341.
[7] Gifford G, Paul J, Vasey PA, Kaye SB, Brown R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin Cancer Res, 2004, 10(13): 4420–4426.
[8] Li D, Kumaraswamy E, Harlan-Williams LM, Jensen RA. The role of BRCA1 and BRCA2 in prostate cancer. Front Biosci (Landmark Ed), 2013, 18(4): 1445–1459.
[9] Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol, 2000, 10(15): 886–895.
[10] Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer, 2012, 12(12): 801–817.
[11] Husain A, He G, Venkatraman ES, Spriggs DR. BRCA1 up-regulation is associated with repair-mediated resistance to cis-diamminedichloroplatinum(II). Cancer Res, 1998, 58(6): 1120–1123.
[12] Kennedy RD, Quinn JE, Mullan PB, Johnston PG, Harkin DP. The role of BRCA1 in the cellular response to chemotherapy. J Natl Cancer Inst, 2004, 96(22): 1659–1668.
[13] Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, Schutte M, Baylin SB, Herman JG. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst, 2000, 92(7): 564–569.
[14] Teodoridis JM, Hall J, Marsh S, Kannall HD, Smyth C, Curto J, Siddiqui N, Gabra H, McLeod HL, Strathdee G, Brown R. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res, 2005, 65(19): 8961–8967.
[15] Tolomeo M, Simoni D. Drug resistance and apoptosis in cancer treatment: development of new apoptosis-inducing agents active in drug resistant malignancies. Curr Med Chem Anticancer Agents, 2002, 2(3): 387–401.
[16] Jones PA. Cancer: death and methylation. Nature, 2001, 409(6817): 141–144.
[17] Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M,Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordon-Cardo C, Lowe SW. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature, 2001, 409(6817): 207–211.
[18] Fulda S, Kufer MU, Meyer E, van Valen F, Dockhorn-Dworniczak B, Debatin KM. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene, 2001, 20(41): 5865–5877.
[19] Chaopatchayakul P, Jearanaikoon P, Yuenyao P, Limpaiboon T. Aberrant DNA methylation of apoptotic signaling genes in patients responsive and nonresponsive to therapy for cervical carcinoma. Am J Obstet Gynecol, 2010, 202(3): 281.e1–281.e9.
[20] Glavinas H, Krajcsi P, Cserepes J, Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr Drug Deliv, 2004, 1(1): 27–42.
[21] Tada Y, Wada M, Kuroiwa K, Kinugawa N, Harada T, Nagayama J, Nakagawa M, Naito S, Kuwano M. MDR1 gene overexpression and altered degree of methylation at the promoter region in bladder cancer during chemotherapeutic treatment. Clin Cancer Res, 2000, 6(12): 4618–4627.
[22] Kusaba H, Nakayama M, Harada T, Nomoto M, Kohno K, Kuwano M, Wada M. Association of 5' CpG demethylation and altered chromatin structure in the promoter region with transcriptional activation of the multidrug resistance 1 gene in human cancer cells. Eur J Biochem, 1999, 262(3): 924–932.
[23] To KK, Zhan Z, Bates SE. Aberrant promoter methylation of the ABCG2 gene in renal carcinoma. Mol Cell Biol, 2006, 26(22): 8572–8585.
[24] Bram EE, Stark M, Raz S, Assaraf YG. Chemotherapeutic drug-induced ABCG2 promoter demethylation as a novel mechanism of acquired multidrug resistance. Neoplasia, 2009, 11(12): 1359–1370.
[25] Ji N, Yuan J, Liu J, Tian S. Developing multidrug-resistant cells and exploring correlation between BCRP/ABCG2 over-expression and DNA methyltransferase. Acta Biochim Biophys Sin (Shanghai), 2010, 42(12): 854–862.
[26] Yao D, Ding S, Burchell B, Wolf CR, Friedberg T. Detoxication of vinca alkaloids by human P450 CYP3A4-mediated metabolism: implications for the development of drug resistance. J Pharmacol Exp Ther, 2000, 294(1): 387–395.
[27] Martinez C, Garcia-Martin E, Pizarro RM, Garcia-Gamito FJ, Agundez JA. Expression of paclitaxel-inactivating CYP3A activity in human colorectal cancer: implications for drug therapy. Br J Cancer, 2002, 87(6): 681–686.
[28] Anttila S, Hakkola J, Tuominen P, Elovaara E, Husgafvel-Pursiainen K, Karjalainen A, Hirvonen A, Nurminen T. Methylation of cytochrome P4501A1 promoter in the lung is associated with tobacco smoking. Cancer Res, 2003, 63(24): 8623–8628.
[29] Habano W, Gamo T, Sugai T, Otsuka K, Wakabayashi G, Ozawa S. CYP1B1, but not CYP1A1, is downregulated by promoter methylation in colorectal cancers. Int J Oncol, 2009, 34(4): 1085–1091.
[30] Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med, 2013, 2(1): 3.
[31] Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer, 2005, 5(4): 275–284.
[32] Schoenhals M, Kassambara A, De Vos J, Hose D, Moreaux J, Klein B. Embryonic stem cell markers expression in cancers. Biochem Biophys Res Commun, 2009, 383(2): 157–162.
[33] Tian T, Zhang Y, Wang S, Zhou J, Xu S. Sox2 enhances the tumorigenicity and chemoresistance of cancer stem-like cells derived from gastric cancer. J Biomed Res, 2012, 26(5): 336–345.
[34] Bareiss PM, Paczulla A, Wang H, Schairer R, Wiehr S, Kohlhofer U, Rothfuss OC, Fischer A, Perner S, Staebler A, Wallwiener D, Fend F, Fehm T, Pichler B, Kanz L, Quintanilla-Martinez L, Schulze-Osthoff K, Essmann F, Lengerke C. SOX2 expression associates with stem cell state in human ovarian carcinoma. Cancer Res, 2013, 73(17): 5544–5555.
[35] Chen YC, Hsu HS, Chen YW, Tsai TH, How CK, Wang CY, Hung SC, Chang YL, Tsai ML, Lee YY, Ku HH, Chiou SH. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS ONE, 2008, 3(7): e2637.
[36] Alonso MM, Diez-Valle R, Manterola L, Rubio A, Liu D, Cortes-Santiago N, Urquiza L, Jauregi P, Lopez de Munain A, Sampron N, Aramburu A, Tejada-Solis S, Vicente C, Odero MD, Bandres E, Garcia-Foncillas J, Idoate MA, Lang FF, Fueyo J, Gomez-Manzano C. Genetic and epigenetic modifications of Sox2 contribute to the invasive phenotype of malignant gliomas. PLoS ONE, 2011, 6(11): e26740.
[37] Zhang HJ, Siu MK, Wong ES, Wong KY, Li AS, Chan KY, Ngan HY, Cheung AN. Oct4 is epigenetically regulated by methylation in normal placenta and gestational trophoblastic disease. Placenta, 2008, 29(6): 549–554.
[38] Wang XQ, Ongkeko WM, Chen L, Yang ZF, Lu P, Chen KK, Lopez JP, Poon RT, Fan ST. Octamer 4 (Oct4) me-diates chemotherapeutic drug resistance in liver cancer cells through a potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology, 2010, 52(2): 528–539.
[39] Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D'Andrea AD. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med, 2003, 9(5): 568–574.
[40] Lee KD, Pai MY, Hsu CC, Chen CC, Chen YL, Chu PY, Lee CH, Chen LT, Chang JY, Huang TH, Hsiao SH, Leu YW. Targeted Casp8AP2 methylation increases drug resistance in mesenchymal stem cells and cancer cells. Biochem Biophys Res Commun, 2012, 422(4): 578–585.
[41] Tian K, Wang Y, Huang Y, Sun B, Li Y, Xu H. Methylation of WTH3, a possible drug resistant gene, inhibits p53 regulated expression. BMC Cancer, 2008, 8: 327.
[42] Nyce J. Drug-induced DNA hypermethylation and drug resistance in human tumors. Cancer Res, 1989, 49(21): 5829–5836.
[43] Kastl L, Brown I, Schofield AC. Altered DNA methylation is associated with docetaxel resistance in human breast cancer cells. Int J Oncol, 2010, 36(5): 1235–1241.
[44] Segura-Pacheco B, Perez-Cardenas E, Taja-Chayeb L, Chavez-Blanco A, Revilla-Vazquez A, Benitez-Bribiesca L, Duenas-Gonzalez A. Global DNA hypermethylationassociated cancer chemotherapy resistance and its reversion with the demethylating agent hydralazine. J Transl Med, 2006, 4: 32.
[45] Shi JF, Li XJ, Si XX, Li AD, Ding HJ, Han X, Sun YJ. ER alpha positively regulated DNMT1 expression by binding to the gene promoter region in human breast cancer MCF-7 cells. Biochem Biophys Res Commun, 2012, 427(1): 47–53.
[46] Cui M, Wen Z, Yang Z, Chen J, Wang F. Estrogen regulates DNA methyltransferase 3B expression in Ishikawa endometrial adenocarcinoma cells. Mol Biol Rep, 2009, 36(8): 2201–2207.
[47] Richards KL, Zhang B, Baggerly KA, Colella S, Lang JC, Schuller DE, Krahe R. Genome-wide hypomethylation in head and neck cancer is more pronounced in HPV-negative tumors and is associated with genomic instability. PLoS ONE, 2009, 4(3): e4941.
[48] Li J, Harris RA, Cheung SW, Coarfa C, Jeong M, Goodell MA, White LD, Patel A, Kang SH, Shaw C, Chinault AC, Gambin T, Gambin A, Lupski JR, Milosavljevic A. Genomic hypomethylation in the human germline associates with selective structural mutability in the human genome. PLoS Genet, 2012, 8(5): e1002692.
[49] Rodriguez J, Frigola J, Vendrell E, Risques RA, Fraga MF, Morales C, Moreno V, Esteller M, Capella G, Ribas M, Peinado MA. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res, 2006, 66(17): 8462–9468.
[50] Gasior SL, Wakeman TP, Xu B, Deininger PL. The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol, 2006, 357(5): 1383–1393.
[51] Daskalos A, Nikolaidis G, Xinarianos G, Savvari P, Cassidy A, Zakopoulou R, Kotsinas A, Gorgoulis V, Field JK, Liloglou T. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int J Cancer, 2009, 124(1): 81–87.
[52] Kazazian HH, Jr, Goodier JL. LINE drive. Retrotransposition and genome instability. Cell, 2002, 110(3): 277–280.
[53] Wang D, Zhou J, Liu X, Lu D, Shen C, Du Y, Wei FZ, Song B, Lu X, Yu Y, Wang L, Zhao Y, Wang H, Yang Y, Akiyama Y, Zhang H, Zhu WG. Methylation of SUV39H1 by SET7/9 results in heterochromatin relaxation and genome instability. Proc Natl Acad Sci USA, 2013, 110(14): 5516–5521.
[54] Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet, 2002, 3(6): 415–428.
[55] Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science, 2003, 300(5618): 455.
[56] Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature, 1998, 396(6712): 643–649.
[57] McClelland SE, Burrell RA, Swanton C. Chromosomal instability: a composite phenotype that influences sensitivity to chemotherapy. Cell Cycle, 2009, 8(20): 3262–3266.
[58] Swanton C, Tomlinson I, Downward J. Chromosomal instability, colorectal cancer and taxane resistance. Cell Cycle, 2006, 5(8): 818–823.
[59] Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IP, Howell M, Kschischo M, Swanton C. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res, 2011, 71(5): 1858–1870.
[60] Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer, 2012, 12(7): 487–493.
[61] Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet, 2009, 10(10): 691–703.
[62] Chalitchagorn K, Shuangshoti S, Hourpai N, Kongruttanachok N, Tangkijvanich P, Thong-ngam D, Voravud N, Sriuranpong V, Mutirangura A. Distinctive pattern of LINE-1 methylation level in normal tissues and theassociation with carcinogenesis. Oncogene, 2004, 23(54): 8841–8846.
[63] Pattamadilok J, Huapai N, Rattanatanyong P, Vasurattana A, Triratanachat S, Tresukosol D, Mutirangura A. LINE-1 hypomethylation level as a potential prognostic factor for epithelial ovarian cancer. Int J Gynecol Cancer, 2008, 18(4): 711–717.
[64] Wanichnopparat W, Suwanwongse K, Pin-On P, Aporntewan C, Mutirangura A. Genes associated with the cis-regulatory functions of intragenic LINE-1 elements. BMC Genomics, 2013, 14: 205.
[65] Miyakura Y, Sugano K, Konishi F, Ichikawa A, Maekawa M, Shitoh K, Igarashi S, Kotake K, Koyama Y, Nagai H. Extensive methylation of hMLH1 promoter region predominates in proximal colon cancer with microsatellite instability. Gastroenterology, 2001, 121(6): 1300–1309.
[66] Leung SY, Yuen ST, Chung LP, Chu KM, Chan AS, Ho JC. hMLH1 promoter methylation and lack of hMLH1 expression in sporadic gastric carcinomas with highfrequency microsatellite instability. Cancer Res, 1999, 59(1): 159–164.
[67] Rice JC, Ozcelik H, Maxeiner P, Andrulis I, Futscher BW. Methylation of the BRCA1 promoter is associated with decreased BRCA1 mRNA levels in clinical breast cancer specimens. Carcinogenesis, 2000, 21(9): 1761–1765.
[68] Cashen AF, Schiller GJ, O'Donnell MR, DiPersio JF. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol, 2010, 28(4): 556–561.
[69] Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C, Ravandi F, Helmer R, Shen L, Nimer SD, Leavitt R, Raza A, Saba H. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer, 2006, 106(8): 1794–1803.
[70] Kaminskas E, Farrell A, Abraham S, Baird A, Hsieh LS, Lee SL, Leighton JK, Patel H, Rahman A, Sridhara R, Wang YC, Pazdur R. Approval summary: azacitidine for treatment of myelodysplastic syndrome subtypes. Clin Cancer Res, 2005, 11(10): 3604–3608.
[71] Oliveira CA, Nie R, Carnes K, Franca LR, Prins GS, Saunders PT, Hess RA. The antiestrogen ICI 182,780 decreases the expression of estrogen receptor-alpha but has no effect on estrogen receptor-beta and androgen receptor in rat efferent ductules. Reprod Biol Endocrinol, 2003, 1: 75.
[72] Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J, Figueroa ME, De Figueiredo Pontes LL, Alberich-Jorda M, Zhang P, Wu M, D'Alo F, Melnick A, Leone G, Ebralidze KK, Pradhan S, Rinn JL, Tenen DG. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature, 2013, 503(7476): 371–376.
(责任编委: 朱卫国)
Aberrant DNA methylation and drug resistance of tumor cells
Xinxin Si, Yujie Sun
Key Laboratory of Human Functional Genomics of Jiangsu Province, Department of Cell Biology, School of Basic Medical Sciences, Nanjing Medical University, Nanjing 210029, China
Drug resistance is one of the major obstacles limiting the success of cancer chemotherapy. The underlying mechanisms are complex. In recent years, the contribution of epigenetic changes to drug resistance has drawn increasing attention. DNA methylation is an important epigenetic modification that plays an important role in regulating gene expression and maintaining genome stability. Primary or acquired resistance of tumor cells is usually accompanied by aberrant DNA methylation. Accumulating evidence has shown that aberrant DNA methylation is involved in drug resistance of tumor cells. In this review, we briefly review the relationship between DNA methylation and drug resistance of tumor cells as well as the underlying mechanisms.
cancer drug resistance; DNA methylation; genome stability; gene expression
2013-10-22;
2014-02-23
国家自然科学基金项目(编号:81172091)和江苏省普通高校研究生科研创新计划项目(编号:CXZZ13_0561)资助
司鑫鑫, 博士研究生, 研究方向:肿瘤耐药与表观遗传。E-mail: sixinxin@njmu.edu.cn
孙玉洁, 教授, 博士生导师, 研究方向:肿瘤耐药与表观遗传。E-mail: yujiesun@njmu.edu.cn
10.3724/SP.J.1005.2014.0411
时间: 2014-3-3 12:41:51
URL: http://www.cnki.net/kcms/detail/11.1913.R.20140303.1241.003.html
猜你喜欢
保健与生活(2022年11期)2022-06-09
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
生物学通报(2020年10期)2020-08-13
天然产物研究与开发(2018年8期)2018-09-10
中成药(2018年7期)2018-08-04
西南国防医药(2016年7期)2016-12-01
国际妇产科学杂志(2016年2期)2016-06-16
癌变·畸变·突变(2015年3期)2015-02-27
