
不同组分下富锂正极材料xLi2MnO3·(1-x)LiNi0.5Mn0.5O2(x=0.1-0.8)的晶体结构与电化学性能
2014-06-23 06:51胡道中苏岳锋李维康王包丽颖
物理化学学报 2014年3期
陈 来 陈 实,2 胡道中 苏岳锋,2,* 李维康王 昭 包丽颖,2 吴 锋,2
(1北京理工大学化工与环境学院,环境科学与工程北京市重点实验室,北京100081;2国家高技术绿色材料发展中心,北京100081;3中国北方车辆研究所,北京100072)
1 引言
不可再生资源日益稀缺,储能技术的发展被认为是实现可持续性发展的关键,1-4关系到国家的能源安全.锂离子电池因其高比能量、高功率、高循环寿命、高安全性和环境友好等优点,5,6成为最理想的储能设备之一,自上世纪末索尼公司将其商品化以来,7其应用领域已从手机、笔记本电脑、相机等便携式电子设备向电动工具、纯电动汽车(EVs)等大功率设备转变,3,8这对锂离子电池的性能,尤其是能量密度与功率密度,提出了更高的要求.9-11
美、日等国对下一代锂离子动力电池体系的能量密度要求已达300 Wh·kg-1,12所以提高正极材料的比容量对构建高比能锂离子电池体系起着决定性作用.13,14传统的正极材料如LiCoO2、LiFePO4、LiMn1/3Ni1/3Co1/3O2等,因较低的比容量(<200 mAh·g-1),难以满足构建高比能锂离子电池体系对正极材料比容量的要求;15而基于Li2MnO3的层状富锂型正极材料,16-21xLi2MnO3·(1-x)LiMO2(0<x<1,M=Ni0.5Mn0.5,Mnx′Niy′Co1-x′-y′,0<x′,y′<1),其可逆容量能达到 250 mAh·g-1以上,22成为最有可能实现300 Wh·kg-1高能量密度锂离子电池体系的正极材料之一.此外,相较其它层状正极材料,较少的钴含量也使其更具成本与环境友好方面的优势.
层状富锂材料xLi2MnO3·(1-x)LiMO2一般认为包含Li2MnO3与LiMO2两种组分,23-25Li2MnO3组分的存在使得该类材料在首次充电大于4.5 V时出现平台,该平台对应于Li2MnO3组分中Li+脱出伴随O 2p键氧化(净脱出形式为Li2O).19,26,27这一现象导致富锂材料发生结构重排,使非电化学活性的Li2MnO3组分活化转变为具有电化学活性的层状材料MnO2,28因而使富锂材料表现出更高的可逆容量.可见,在富锂材料xLi2MnO3·(1-x)LiMO2中,Li2MnO3组分有着举足轻重的作用,当其含量发生变化时,富锂材料的电化学性能与结构也会随之改变;16,29因此,研究不同组分含量下,富锂材料xLi2MnO3·(1-x)LiMO2的结构与电化学性能变化有着重要意义,对进一步改善其电化学性能具有指导作用.基于这个目的,本文采用溶胶-凝胶法,30-32以柠檬酸为螯合剂,制备了一系列富锂材料xLi2MnO3·(1-x)LiNi0.5Mn0.5O2(x=0.1-0.8,如图1所示),并对其进行了物理和电化学性能表征,以期了解不同组分含量下富锂材料的结构与电化学性能.
2 实验部分
2.1 样品的制备
合成原料为国药集团化学试剂有限公司生产的 CH3COOLi·2H2O(分析纯,99%)、(CH3COO)2Ni·4H2O(分析纯,98%)、(CH3COO)2Mn·4H2O(分析纯,99%)、C6H8O7·H2O(分析纯,99.5%).根据目标产物化学计量比取适量的乙酸锂(过量5%以弥补高温煅烧时的损失)、乙酸镍、乙酸锰,与适量的柠檬酸分别溶解于去离子水中后,将柠檬酸溶液逐滴加入到过渡金属盐溶液中不断搅拌;滴加完成后用稀氨水调节混合液的pH值稳定在8-9之间;后将溶液置于90-100°C的水浴中蒸发至绿色凝胶状,于真空干燥箱中80°C干燥12 h后取出压成圆片,转移至马弗炉中450°C煅烧5 h,冷却后,将圆片研磨至细粉状,再次压成圆片,置于马弗炉中900°C煅烧24 h,随炉冷却后取出研磨至细粉状,得最终产物xLi2MnO3·(1-x)LiNi0.5Mn0.5O2.
2.2 结构和形貌的表征
采用日本Rigaku公司生产的IV-185型旋转阳极衍射仪进行晶体结构的表征,测试条件为:Cu靶,Kα射线,靶电压40 kV,靶电流40 mA,扫描速率为8(°)·min-1,扫描范围为10°-90°.采用荷兰FEI公司生产的QUANTA6000扫描电镜进行材料的形貌分析.
2.3 电化学性能测试
活性正极材料、乙炔黑(日本DENKA生产)、聚偏氟乙烯(PVDF,有色金属研究总院)以8:1:1的质量比调制成浆料,用N-甲基-2-吡咯烷酮(NMP)调至适当粘度后均匀涂于集流体上,置于80°C的烘箱中干燥12 h后裁成电极极片并用分析天平称重待用.扣式电池的组装在氩气手套箱内进行,以金属锂片作负极,Celgard 2300多孔复合聚合物膜为隔膜,1 mol·L-1LiPF6的碳酸乙烯酯 (EC)、碳酸二甲酯(DMC)(体积比1:1)的混合溶液作为电解液,组装成CR2025型扣式电池,静置24 h后,采用武汉金诺公司生产的LAND电池测试系统于室温下进行恒电流充放电循环测试,充放电区间为2.0-4.8 V,充放电电流以1C=250 mA·g-1计算.

图1 xLi2MnO3·(1-x)LiNi0.5Mn0.5O2(x=0.1-0.8)组成示意图Fig.1 Schematic diagram of xLi2MnO3·(1-x)LiNi0.5Mn0.5O2(x=0.1-0.8)
3 结果与讨论
3.1 不同组分对材料形貌的影响
图2为x为不同值时所制备的材料的SEM图,放大倍数均为50000倍.图中可见,除了x=0.6时所制备的材料颗粒稍大,其它x值下的材料在形貌上并无太大的区别,所得颗粒均匀但呈无规则团聚状,一次粒子粒径较小,均在准微米尺度.由于所制备的样品采用了同样的合成方法与煅烧工艺,因此尽管x的取值从0.1到0.8大幅变化,也未过多影响最终产物的形貌;由此可见,材料组分大幅变化时并未对产物的形貌产生影响.
3.2 不同组分对材料结构的影响
层状富锂材料xLi2MnO3·(1-x)LiNi0.5Mn0.5O2包含的两种组分Li2MnO3与LiNi0.5Mn0.5O2中,LiNi0.5Mn0.5O2具有与LiCoO2相同的α-NaFeO2型层状结构,33属于六方晶系,R3m空间点阵群;而在另一组分Li2MnO3中,过量的Li+在过渡金属层中与Mn4+以1:2的比例占据α-NaFeO2中的Fe3+位,形成LiMn6超结构,18致其晶格对称性较与LiNi0.5Mn0.5O2有所下降,34-36但仍具有由α-NaFeO2衍生而来的层状结构.因此正如图3中所示,所合成的富锂材料的X射线衍射峰与LiCoO2峰位对应较好,尤其是(006)/(012)、(018)/(110)两对峰分裂明显,表明具有良好的层状结构.
Li+在Li2MnO3组分过渡金属层中形成的LiMn6超结构在XRD表征中表现为2θ=20°-25°区域内微弱的超晶格衍射峰,37,38这一结构特点在所制备的材料中均得到了体现(图中虚线框所示),且随着Li2MnO3组分的增多,2θ=20°-25°处微弱的衍射峰峰强相应增大.Li2MnO3有别于LiNi0.5Mn0.5O2的这一结构特点,使得两者相对含量发生变化时,对所合成的富锂材料的结构产生了影响.当x值较小时(如x=0.1-0.3),相应材料在2θ=36.5°,38.0°,44.2°,64.4°等处出现了杂质峰(图中*号所示处),通过与图3底部两种尖晶石相标准PDF卡峰位的对比发现,这些杂质峰与尖晶石相Li4Mn5O12(Fd3m)、LiNi0.5Mn1.5O4(P4332)对应均较好,其归属难以判断,但可以确定的是,这些杂质峰来自于尖晶石相,这一发现与Amine等39的报道略有不同;此外,通过图3中A、B图对比可见,当x值增大时,这些杂质峰逐渐变弱直至消失,这表明,Li2MnO3组分较多时能有效抑制尖晶石杂相的生成.
表1列出了x=0.4-0.8的材料基于α-NaFeO2结构计算所得的晶格参数与特征峰强度比(x=0.1-0.3的样品由于杂质峰的干扰未做计算),各样品的c/a值均大于4.899,说明材料均具备良好的层状结构;40,41同时,随着x的增大,I(003)/I(104)峰强比值不断增大.对于层状结构的材料,I(003)/I(104)的比值常被用来表征阳离子混排的程度,当I(003)/I(104)>1.2时,说明材料的阳离子混排程度较低.42阳离子混排对材料的电化学性能有较大的影响,主要是因为混入Li位的Ni2+在充电过程中氧化为离子半径更小的Ni3+后,易导致结构坍塌,阻碍Li+的脱嵌,影响材料的电化学性能.43在xLi2MnO3·(1-x)LiMn0.5Ni0.5O2层状富锂材料中,Ni2+与Li+的离子半径较为接近是发生阳离子混排的主要原因;44,45当Li2MnO3组分增多时,材料中的Ni2+含量相应减少,阳离子混排程度随之降低.

图2 不同xLi2MnO3·(1-x)LiNi0.5Mn0.5O2组分下材料的扫描电镜(SEM)图Fig.2 Scanning electron microscopy(SEM)images of the materials with different x in xLi2MnO3·(1-x)LiNi0.5Mn0.5O2

图3 不同x值下材料的X射线衍射(XRD)图Fig.3 Powder X-ray diffraction patterns of the materials with different x
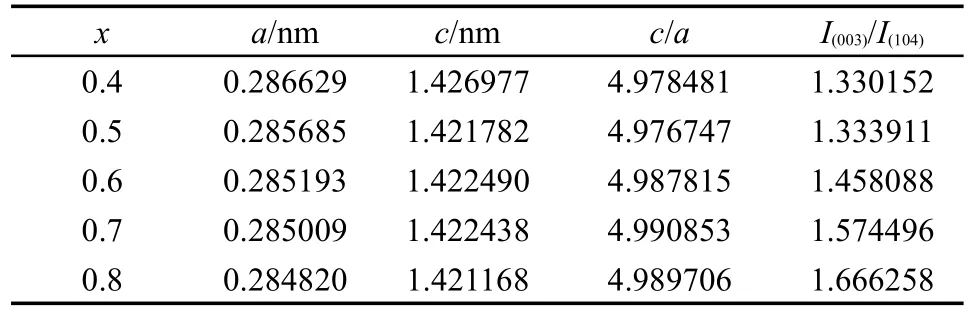
表1 不同x值下材料的晶胞参数与峰强比Table 1 Lattice parameters and ratio of characteristic peak intensities of the materials with different x
3.3 不同组分对材料电化学性能的影响
图4为不同x值下样品在0.1C(25 mA·g-1)下的首次充放电曲线图,具体数值列于表2中;图中xy平面上的红色投影线为各样品首次充电过程中电压大于4.5 V时所对应的充电容量,绿色投影线为电压小于4.5 V时所对应的充电容量.从图4中可以清楚地看到,除了x=0.8的样品,随着x的增大,材料首次充电容量也随之增大.
xLi2MnO3·(1-x)LiMn0.5Ni0.5O2的首次充电过程可分为两个阶段,以x=0.5的样品为例(图5),当终止电压在4.5 V以下时,Li+的脱嵌伴随Ni2+/4+的氧化还原反应进行,即图5中a点以左部分;46,47当终止电压在4.5 V以上时,则出现一个较长的平台,48-51即图5中a点以右的部分,该平台对应于富锂材料的电化学活化过程.19,27,28从图4中可以看到,每个样品均出现了层状富锂材料的这一特征平台,与XRD显示的材料结构相吻合.由图5可见,这一现象仅出现于首周充电过程,在随后的充电过程中不再出现这一平台.Johnson等52认为,当电压大于4.5 V时,所对应的电化学反应(式1)为:
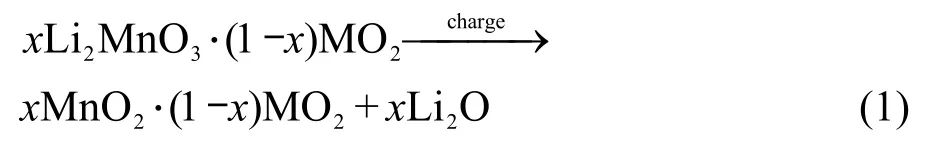
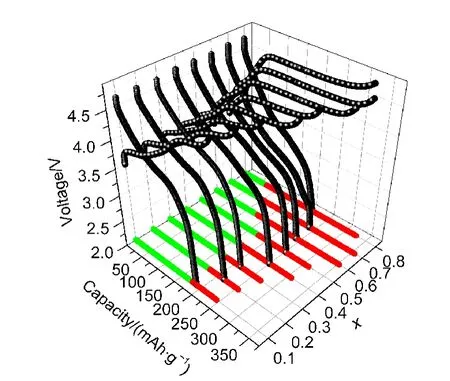
图4 不同x值下xLi2MnO3·(1-x)LiNi0.5Mn0.5O2材料的首次充放电曲线图Fig.4 Initial charge-discharge curves of the materials with different x in xLi2MnO3·(1-x)LiNi0.5Mn0.5O2
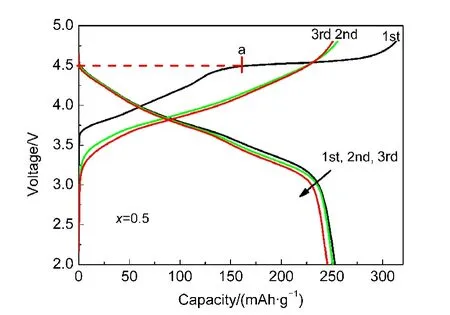
图5 0.5Li2MnO3·0.5LiMn0.5Ni0.5O2前三周的充放电曲线Fig.5 Charge-discharge curves of 0.5Li2MnO3·0.5LiMn0.5Ni0.5O2in the first three cycles
即电压大于4.5 V后,其充电容量来自于Li2MnO3组分的脱锂过程,因此,当x升高时,4.5 V以上的首次充电容量应增大,这与图4中红色投影线所示实验结果一致.
根据Johnson等52的观点,当电压小于4.5 V时,所对应的电化学反应(式(2))为:
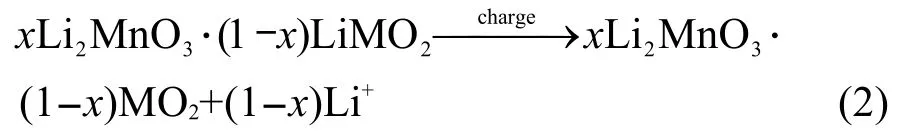
该反应伴随着Ni2+/4+的氧化还原反应,我们将各个样品的首次充电容量(以4.5 V为节点分为两段)与首次放电容量并列于表2中,实验结果与Dahn小组16,17的研究成果相符.由于xLi2MnO3·(1-x)LiMn0.5Ni0.5O2中,Mn为4+,没有电化学活性,53,54因而一般认为式(1)的反应是不可逆的,这也是表2中随着x值的增大,首次库仑效率降低的原因;而式(2)所对应的Ni2+/4+的氧化还原反应则是可逆的,在此假设基础上,我们将首次放电容量减去Ni2+/4+还原反应所提供的容量后发现,首次放电容量超出了Ni4+还原到Ni2+所能提供的容量,两者所相差的容量被命名为“额外的容量”列于表2中.由于xLi2MnO3·(1-x)LiMn0.5Ni0.5O2中,仅有Ni和Mn两种过渡金属元素,因此这部分容量来自于锰离子的变价反应,这说明在经历首次充电过程之后,锰离子得到活化,具备了电化学活性;此外,从表2中也可以看到,随着Li2MnO3组分含量的增大,Mn活化后贡献的容量也呈现上升的趋势.
为了更清楚地了解材料在经历首次充电过程后的变化,作材料前三周循环的容量微分曲线,如图6所示.在第一周充电过程中,3.75与4.2 V左右的氧化峰分别对应于Ni2+/3+与Ni3+/4+的氧化反应,15随着x的增大,LiMn0.5Ni0.5O2组分相对含量逐渐减少,因而容量微分曲线中,3.75与4.2 V左右的氧化还原峰逐渐变得平缓;4.5 V左右出现一个巨大的氧化峰,对应于式(1)的反应,为Li+脱出并伴随氧释放的过程,27如图6中插图所示,随着x的增大,该处的峰逐渐尖锐,除了x=0.8的样品,其它样品在4.5 V左右的氧化峰在随后两周的循环过程中不再出现,印证了其不可逆性,在x=0.8时,如图中虚线框所示,在第二周充电过程中4.5 V处仍出现了微弱的氧化峰,说明该样品在第一周充电过程中并未反应完全,这也解释了表2中样品首次充电容量随x增大而增大,但x=0.8时并不符合这一规律的现象.首次放电过程中,4.2与3.6 V左右的还原峰来自Ni4+/3+与Ni3+/2+的还原反应.15经过首次充电过程后,3.5 V以下出现了一对新的氧化还原峰(如图6中虚线以左所示),该氧化还原峰即来自Li2MnO3活性后产生的氧化还原反应,29,47这表明层状富锂材料xLi2MnO3·(1-x)LiMn0.5Ni0.5O2在经历了首次充放电循环后,Li2MnO3组分得到活化,转变为具有电化学活性的MnO2,并在随后的循环中贡献了部分可逆容量;且随着x的增大,该处的峰越来越明显.
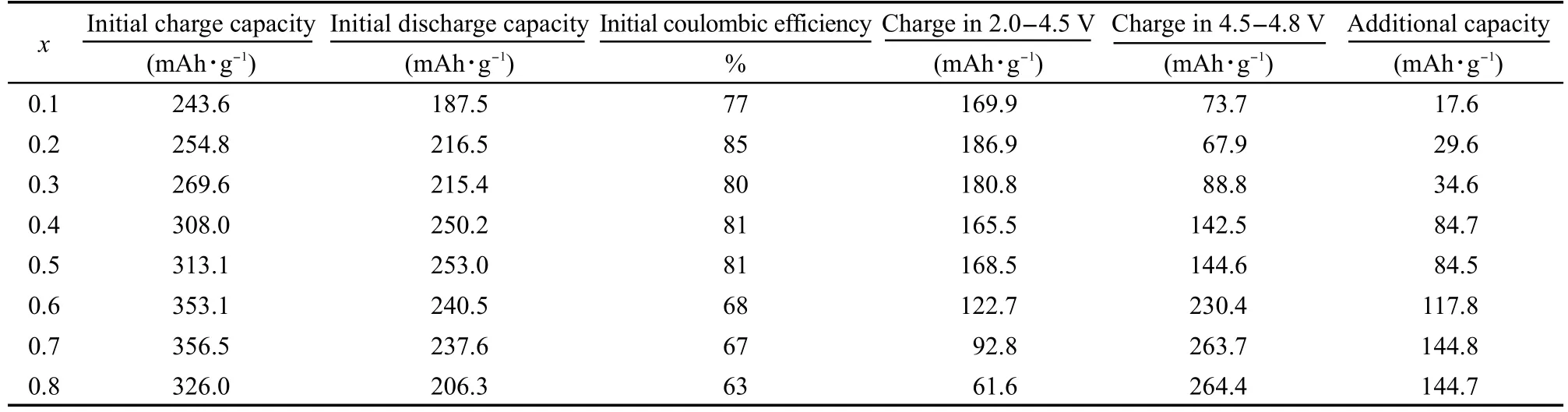
表2 xLi2MnO3·(1-x)LiNi0.5Mn0.5O2中不同x值下材料的电化学数据Table 2 Electrochemical data of the materials with different x in xLi2MnO3·(1-x)LiNi0.5Mn0.5O2

图6 xLi2MnO3·(1-x)LiMn0.5Ni0.5O2(x=0.1-0.8)前三周的容量微分曲线图Fig.6 Differential capacity vs voltage plots of xLi2MnO3·(1-x)LiMn0.5Ni0.5O2(x=0.1-0.8)in the first three cycles
此外,尽管在XRD分析中,在x=0.1-0.3时,材料中出现了尖晶石杂相,但在容量微分曲线中,并未出现明显的Li4Mn5O12或LiNi0.5Mn1.5O2的氧化还原反应峰,55,56这说明尖晶石杂相在整体材料中的含量较少,对容量影响较小,表2中所示容量主要来自于富锂材料.
图7为不同x值下材料0.1C(25 mA·g-1)下40周内的循环曲线.从图中可以看到,当x较低(如x=0.1,0.2)时,其放电容量始终低于其它x较大的样品,但其循环稳定性较好,40周循环内容量保持率较高;而x较大(如x=0.7,0.8)时,尽管前几周的放电容量较高,但在随后的循环过程中容量衰减较快.
材料结构的稳定性对其循环性能有重要影响,Ammundsen等57曾用Li2MnO3来稳定LiCrO2,所制备的材料具有较好的循环稳定性;xLi2MnO3·(1-x)LiMn0.5Ni0.5O2中,Mn4+同样能够稳定材料的结构,58提高其循环性能,但根据之前的分析,当材料经历首次充放电循环后,Mn4+得到活化,在循环过程中发生Mn3+/4+氧化还原反应,Mn不再保持4+;同时,随着Li2MnO3组分含量的增大,Mn4+活化后提供的容量也随之增加(见表2),这意味着更多的Mn4+得到活化,这可能是Li2MnO3组分较多时循环性能较差的原因.
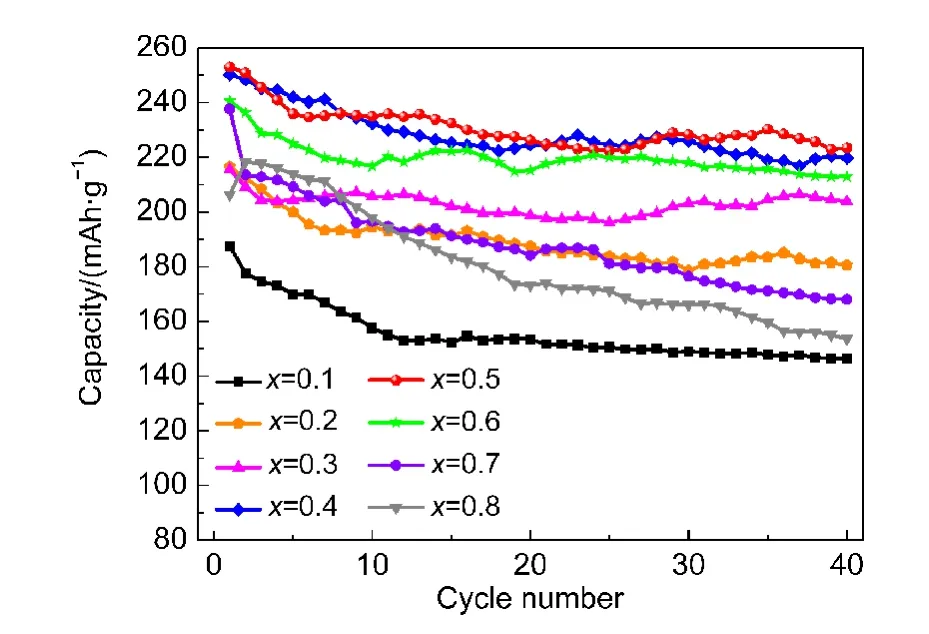
图7 xLi2MnO3·(1-x)LiNi0.5Mn0.5O2中不同x值下材料的循环性能Fig.7 Cycle performance of the materials with different x in xLi2MnO3·(1-x)LiNi0.5Mn0.5O2

图8 0.1C 倍率下0.5Li2MnO3·0.5LiMn0.5Ni0.5O2多次循环后的容量微分曲线图Fig.8 Differential capacity vs voltage plots of the 0.5Li2MnO3·0.5LiMn0.5Ni0.5O2cycle at 0.1C rate
此外,以x=0.5的样品为例,如图8所示作其循环多次后的容量微分曲线可见,经过多次循环后,材料在3.5 V以下由于Li2MnO3活化后引起的氧化还原反应,其氧化还原电位向低电压方向偏移(如图8中箭头所示,其插图为2.8-3.8 V范围下局部放大图),这意味着材料由层状结构逐渐向尖晶石相转变,这也可能是导致循环性能变差的原因;由于Li2MnO3组分增大时,材料中锰的含量增大,更易引起这种转变,因此Li2MnO3组分增加时,循环性能可能恶化,这与实验结果相符.
综上所述,Li2MnO3组分较多时材料循环性能较差,可能是因为材料中有更多的Mn得到活化,不再保持4+;以及在循环过程中层状材料向尖晶石结构转变引起的.同时,Li2MnO3组分较少时,尽管循环更为稳定,放电容量却不高;x=0.4,0.5,0.6时的材料则兼顾了容量高与循环稳定性好的优点.这说明,当Li2MnO3组分含量不是很高时,并不会对循环性能产生太大的影响,并且又能保证较高的放电容量.
4 结论
以溶胶-凝胶法成功合成了一系列富锂锰基正极材料xLi2MnO3·(1-x)LiNi0.5Mn0.5O2(x=0.1-0.8),所用的溶胶-凝胶法具有纯度高、合成时间短、产物粒径小、均一性好等诸多优点,46,59易实现分子水平上的均匀混合,相比固相法等合成方法,较大程度地减少了合成工艺对实验结果的影响;其缺点在于合成过程中由溶胶变为凝胶的过程需要较大的能耗,且对环境有一定影响,因而该合成方法更适用于少量合成.实验结果表明,富锂材料中的两种组分相对含量发生变化时对材料的结构与电化学性能有重大影响:当Li2MnO3组分含量较多时,能有效抑制尖晶石杂相的生成,使富锂材料形成更为完善的层状结构,但其深层机理还需进一步研究;Li2MnO3组分赋予了富锂材料xLi2MnO3·(1-x)LiNi0.5Mn0.5O2突出的首次充电容量,并且在此后的循环过程中,Li2MnO3转变为具有电化学活性的MnO2,提供了部分放电容量,这部分容量随着Li2MnO3组分的增多而增多,与此同时,相对含量减少的LiNi0.5Mn0.5O2组分所提供的放电容量在降低.结果表明,当x=0.5时,材料的首次放电容量最高;此外,Li2MnO3组分还对材料的循环性能产生了重大影响,当Li2MnO3组分较高时,材料的循环性能较差,这可能是因为Li2MnO3组分的存在使得富锂材料xLi2MnO3·(1-x)LiNi0.5Mn0.5O2在循环过程中更易向尖晶石相转变引起的.综合放电容量与循环性能来看,当x=0.5时,材料的电化学性能最优,x=0.4,0.6时材料亦表现出较好的电化学性能,值得关注.本文制备的富锂材料xLi2MnO3·(1-x)LiNi0.5Mn0.5O2组分变化较广,对该材料的性能筛选较为全面,实验结果对进一步改善该材料电化学性能有一定的指导意义,但深层机理方面仍需进一步研究,尤其是Li2MnO3组分引起的材料结构上的变化与电化学性能之间的深层联系值得关注.
(1) Choi,N.S.;Chen,Z.H.;Freunberger,S.A.;Ji,X.L.;Sun,Y.K.;Amine,K.;Yushin,G.;Nazar,L.F.;Cho,J.;Bruce,P.G.Angew.Chem.Int.Edit.2012,51(40),9994.doi:10.1002/anie.201201429
(2) Li,H.;Wang,Z.X.;Chen,L.Q.;Huang,X.J.Adv.Mater.2009,21(45),4593.doi:10.1002/adma.v21:45
(3) Jeong,G.J.;Kim,Y.U.;Kim,H.S.;Kim,Y.J.;Sohn,H.J.Energ Environ.Sci.2011,4(6),1986.doi:10.1039/c0ee00831a
(4) He,P.;Yu,H.J.;De,L.;Zhou,H.S.J.Mater.Chem.2012,22(9),3680.doi:10.1039/c2jm14305d
(5) Whittingham,M.S.Chem.Soc.Rev.2004,104(10),4271.
(6) Aricò,A.S.;Bruce,P.;Scrosati,B.;Tarascon,J.M.;Schalkwijk,W.V.Nat.Mater.2005,4(5),366.doi:10.1038/nmat1368
(7) Nagaura,T.;Tozawa,K.Prog.Batteries Solar Cells1990,9,209.
(8) Song,H.K.;Lee,K.T.;Kim,M.G.;Nazar,L.F.;Cho,J.Adv.Funct.Mater.2010,20(22),3818.doi:10.1002/adfm.201000231
(9) Armand,M.;Tarascon,J.M.Nature2008,451(7179),652.doi:10.1038/451652a
(10) Goodenough,J.B.;Kim,Y.J.Power Sources2011,196(16),6688.doi:10.1016/j.jpowsour.2010.11.074
(11) Wu,F.;Li,N.;Su,Y.F.;Shou,H.F.;Bao,L.Y.;Yang,W.;Zhang,L.J.;An,R.;Chen,S.Adv.Mater.2013,25(27),3722.doi:10.1002/adma.v25.27
(12) Du,K.;Hu,G.R.Chin.Sci.Bull.2012,57(10),794.[杜柯,胡国荣.科学通报,2012,57(10),794.]doi:10.1360/972011-610
(13) Ritchie,A.G.J.Power Sources2001,96(1),1.doi:10.1016/S0378-7753(00)00673-X
(14) Fergus,J.W.J.Power Sources2010,195(4),939.doi:10.1016/j.jpowsour.2009.08.089
(15)Wu,F.;Li,N.;Su,Y.;F.;Lu,H.Q.;Zhang,L.J.;An,R.;Wang,Z.;Bao,L.Y.;Chen,S.J.Mater.Chem.2012,22(4),1489.doi:10.1039/c1jm14459f
(16) Lu,Z.H.;MacNeil,D.D.;Dahn,J.R.Electrochem.Solid St.2001,4(11),A191.
(17) Lu,Z.H.;Beaulieu,L.Y.;Donaberger,R.A.;Thomas,C.L.;Dahn,J.R.J.Electrochem.Soc.2002,149(6),A778.
(18) Thackeray,M.M.;Kang,S.H.;Johnson,C.S.;Vaughey,J,T.;Benedek,R.B.;Hackney,S.A.J.Mater.Chem.2007,17(30),3112.doi:10.1039/b702425h
(19)Armstrong,A.R.;Holzapfel,M.;Novák,P.;Johnson,C.S.;Kang,S.H.;Thackeray,M.M.;Bruce,P.G.J.Am.Chem.Soc.2006,128(26),8694.doi:10.1021/ja062027+
(20)Wang,D.P.;Belharouak,I.;Zhou,G.W.;Amine,K.Adv.Funct.Mater.2013,23(8),1070.doi:10.1002/adfm.v23.8
(21) Chen,Z.;Ren,Y.;Lee,E.;Johnson,C.;Qin,Y.;Amine,K.Adv.Energy Mater.2013,3(6),729.doi:10.1002/aenm.201201059
(22)Park,Y.J.;Hong,Y.S.;Wu,X.L.;Kim,M.G.;Ryu,K.S.;Chang,S.H.J.Electrochem.Soc.2004,151(5),A720.
(23) Meng,Y.S.;Ceder,G.;Grey,C.P.;Yoon,W.S.;Jiang,M.;Bréger,J.;Horn,Y.S.Chem.Mater.2005,17(9),2386.doi:10.1021/cm047779m
(24) Bréger,J.;Jiang,M.;Dupré,N.;Meng,Y.S.;Horn,Y.S.;Ceder,G.;Grey,C.P.J.Solid State Chem.2005,178(9),2575.doi:10.1016/j.jssc.2005.05.027
(25) Kim,J.S.;Johnson,C.S.;Vaughey,J.T.;Thackeray,M.M.Chem.Mater.2004,16(10),1996.doi:10.1021/cm0306461
(26)Armstrong,A.R.;Bruce,P.G.Electrochem.Solid.St.2004,7(1),A1.
(27) Park,M.S.;Lee,J.W.;Choi,W.;Im,D.;Doo,S.G.;Park,K.S.J.Mater.Chem.2010,20(34),7208.doi:10.1039/c0jm00617c
(28)Weill,F.;Tran,N.;Martin,N.;Croguennec,L.;Delmas,C.Electrochem.Solid.St.2007,10(8),A194.
(29) Lu,Z.;Dahn,J.R.J.Electrochem.Soc.2002,149(7),A815.
(30) Park,Y.J.;Hong,Y.S.;Wu,X.;Ryu,K.S.;Chang,S.H.J.Power Sources2004,129(2),288.doi:10.1016/j.jpowsour.2003.11.024
(31)Hong,Y.S.;Park,Y.J.;Ryu,K.S.;Chang,S.H.;Kim,M.G.J.Mater.Chem.2004,14(9),1424.doi:10.1039/b311888f
(32) Jain,G.;Yang,J.S.;Balasubramanian,M.;Xu,J.J.Chem.Mater.2005,17(15),3850.doi:10.1021/cm0503329
(33) Thackeray,M.M.;Kang,S.H.;Johnson,C.S.;Vauqhey,J.T.;Benedek,R.;Hackney,S.A.J.Mater.Chem.2007,17(30),3112.doi:10.1039/b702425h
(34) Bareno,J.;Lei,C.H.;Wen,J.G.;Kang,S.H.;Petrov,I.;Abraham,D.P.Adv.Mater.2010,22(10),1122.doi:10.1002/adma.200904247
(35) Koyama,Y.;Tanaka,I.;Nagao,M.;Kanno,R.J.Power Sources2009,189(1),798.doi:10.1016/j.jpowsour.2008.07.073
(36) Bréger,J.;Meng,Y.S.;Hinuma,Y.;Kumar,S.D.;Kang,K.;Horn,Y.S.;Ceder,G.;Grey,C.P.Chem.Mater.2006,18(20),4768.doi:10.1021/cm060886r
(37) Johnson,C.S.;Li,N.;Lefief,C.;Vaughey,J.T.;Thackeray,M.M.Chem.Mater.2008,20(19),6095.doi:10.1021/cm801245r
(38) Thackeray,M.M.;Johnson,C.S.;Vaughey,J.T.;Li,N.;Hackney,S.A.J.Mater.Chem.2005,15(23),2257.doi:10.1039/b417616m
(39) Belharouak,I.;Koenig,G.M.,Jr.;Ma,J.W.;Wang,D.P.;Amine,K.Electrochem.Commun.2011,13(3),232.doi:10.1016/j.elecom.2010.12.021
(40) Myung,S.T.;Kumagai,N.;Komaba,S.;Chung,H.T.Solid State Ionics2001,139(1),47.
(41) Kim,Y.;Hyun,S.K.;Martin,S.W.Electrochim.Acta2006,52(3),1316.doi:10.1016/j.electacta.2006.07.033
(42) Morales,J.;Perez-Vicente,C.;Tirado,J.L.Mater.Res.Bull.1990,25(5),623.doi:10.1016/0025-5408(90)90028-Z
(43) Delmas,C.;Peres,J.P.;Rougier,A.;Demourgues,A.;Weill,F.;Chadwick,A.;Broussely,M.;Perton,F.;Biensan,P.;Willmann,P.J.Power Sources1997,68(1),120.doi:10.1016/S0378-7753(97)02664-5
(44) Kim,J.M.;Chung,H.T.Electrochim.Acta2004,49(21),3573.doi:10.1016/j.electacta.2004.03.025
(45) Fey,G.T.K.;Shiu,R.F.;Subramanian,V.;Chen,J.G.;Chen,C.L.J.Power Sources2002,103(2),265.doi:10.1016/S0378-7753(01)00859-X
(46) Kim,J.H.;Park,C.W.;Sun,Y.K.Solid State Ionics2003,164(1),43.
(47) Yu,L.;Qiu,W.;Lian,F.;Huang,J.;Kang,X.L.J.Alloy.Compd.2009,471(1),317
(48) Rossouw,M.H.;Liles,D.C.;Thackeray,M.M.J.Solid State Chem.1993,104(2),464.doi:10.1006/jssc.1993.1182
(49)Robertson,A.D.;Bruce,P.G.Chem.Commun.2002,2790.
(50) Tran,N.;Croguennec,L.;Ménétrier,M.;Weill,F.;Biensan,P.;Jordy,C.;Delmas,C.Chem.Mater.2008,20(15),4815.doi:10.1021/cm070435m
(51) Jiang,M.;Key,B.;Meng,Y.S.;Grey,C.P.Chem.Mater.2009,21(13),2733.doi:10.1021/cm900279u
(52) Johnson,C.S.;Li,N.;Lefief,C.;Thackeray,M.M.Electrochem.Commun.2007,9(4),787.doi:10.1016/j.elecom.2006.11.006
(53) Kalyani,P.;Chitra,S.;Mohan,T.;Gopukumar,S.J.Power Sources1999,80(1),103.
(54) Robertson,A.D.;Bruce,P.G.Chem.Mater.2003,15(10),1984.doi:10.1021/cm030047u
(55) Toshimi,T.;Hiroshi,H.;Etsuo,A.K.J.Solid State Chem.1995,115(2),420.doi:10.1006/jssc.1995.1154
(56) Belharouak,I.;Koenig,G.M.,Jr.;Ma,J.W.;Wang,D.P.;Amine,K.Electrochem.Commun.2011,13(3),232.doi:10.1016/j.elecom.2010.12.021
(57)Ammundsen,B.;Paulsen,J.Adv.Mater.2001,13(12-13),943.doi:10.1002/1521-4095(200107)13:12/13<943::AIDADMA943>3.0.CO;2-J
(58) Johnson,C.S.;Korte,S.D.;Vaughey,J.T.;Thackeray,M.M.;Bofinger,T.E.;Hom,Y.S.;Hackney,S.A.J.Power Sources1999,81,491.
(59)Wang,Z.;Wu,F.;Su,Y.F.;Bao,L.Y.;Chen,L.;Li,N.;Chen,S.Acta Phys.-Chim.Sin.2012,28(4),823.[王 昭,吴 锋,苏岳锋,包丽颖,陈 来,李 宁,陈 实.物理化学学报,2012,28(4),823.]doi:10.3866/PKU.WHXB201202102
猜你喜欢
煤气与热力(2021年12期)2022-01-19
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
重型机械(2019年3期)2019-08-27
表面工程与再制造(2019年6期)2019-08-24
中成药(2018年8期)2018-08-29
中成药(2018年2期)2018-05-09
资源节约与环保(2018年1期)2018-02-08
焊接(2016年9期)2016-02-27
新疆钢铁(2015年2期)2015-11-07
