
以痉挛性截瘫为主要表现且不伴有脑白质异常信号的肾上腺脑白质营养不良的误诊分析(附3例报告)
2017-11-03 12:39卢岩刘艳秋张新卿矫黎东高乐虹王宪玲
临床神经病学杂志 2017年5期
卢岩,刘艳秋,张新卿,矫黎东,高乐虹,王宪玲
·学术交流·
以痉挛性截瘫为主要表现且不伴有脑白质异常信号的肾上腺脑白质营养不良的误诊分析(附3例报告)
卢岩,刘艳秋,张新卿,矫黎东,高乐虹,王宪玲
目的探讨以痉挛性截瘫为主要表现且不伴有脑白质异常信号的肾上腺脑白质营养不良(ALD)的临床特点,以免误诊为遗传性痉挛性截瘫(HSP)。方法回顾性分析3例曾误诊为HSP的经基因检测确诊的以痉挛性截瘫为主要表现且不伴有脑白质异常信号的ALD患者的临床资料。结果3例患者均隐袭起病,缓慢进展,临床表现为显著的痉挛性截瘫,伴或不伴自主神经或周围神经损害,影像学均未见脑白质异常信号,临床诊断均怀疑为HSP,后经基因检测确诊为肾上腺脊髓神经病(AMN)。基因测序结果分别为ABCD1基因exon 2的c.961_963del缺失突变,ABCD1基因exon 1的c.310C>T错义突变,ABCD1基因exon 3的c.1202G>A错义突变。例1及例3家系验证示,二者母亲均存在相应基因突变。结论AMN是ALD的一种亚型,以双下肢痉挛性截瘫为主要表现,不伴有脑白质异常信号的患者易被误诊为HSP。因此,对临床上怀疑HSP的患者,尤其是男性患者,应进行极长链脂肪酸筛查,有条件者应行基因检测,以鉴别ALD。
肾上腺脑白质营养不良;肾上腺脊髓神经病;遗传性痉挛性截瘫;基因检测;鉴别诊断
肾上腺脑白质营养不良(ALD)是最常见的溶酶体病,呈X连锁隐性遗传,根据发病年龄、受累部位、进展缓急等共分为7种亚型[1]。肾上腺脊髓神经病(AMN)作为ALD的一种临床表型,约占40%~46%,发病年龄为19~37岁,主要表现为脊髓受累的症状,包括痉挛性截瘫、下肢深感觉障碍以及排尿障碍等,可伴或不伴周围神经损伤,上肢不受累或轻度受累,病程可达10年或更长[2]。根据伴或不伴脑部病变,可将AMN分为两类,分别为单纯型AMN 与伴有脑部病变的AMN[1]。约60%AMN患者的头颅MRI可不伴有脑白质异常信号,仅表现为脑干或内囊中皮质脊髓束的华勒变性[2]。遗传性痉挛性截瘫(HSP)是以双下肢痉挛性瘫痪为主要表现的神经系统退行性疾病,可分为单纯型和复杂型[3]。临床上,以痉挛性瘫痪为主要表现且不伴有脑白质异常信号的AMN患者常常被误诊为HSP。本研究回顾性分析了2014~2015年我院病房收治的3例以痉挛性截瘫为主要表现的AMN患者的临床资料,3例患者均曾被误诊为HSP,后经基因检测确诊为AMN。现总结这3例患者的临床表现及影像学特征,以寻找HSP与ALD的鉴别点,为临床诊断提供帮助。
1 临床资料
1.1 例1 男,25岁,主因“双下肢无力4年,加重5个月”于2015年9月入院。患者于4年前无明显诱因出现双下肢无力,跑步及跨越障碍物时明显,不影响行走。2年前患者自觉双下肢僵硬,于当地医院查头颈胸腰段MRI均未见明显异常。5个月前患者双下肢无力、僵硬较前加重,无肌肉萎缩,无深浅感觉障碍,双上肢活动无异常。为求进一步诊治于我院就诊。无家族史,病后体质量无减轻。入院体检:神清、语利。高级皮质功能及颅神经检查均未见异常。双上肢肌力、肌张力、腱反射正常,双下肢肌张力增高,肌力Ⅴ-级,腱反射()。深浅感觉查体未见明显异常。双侧Hoffmann征、Babinski征阳性。指鼻试验、跟-膝-胫试验稳准。入院后完善检查:血尿便常规、肝肾功能、甲状腺功能、叶酸、维生素B12、抗核抗体谱等均未见明显异常。CSF常规、生化、免疫、病毒等均正常。头MRI未见明显异常。EMG示上下肢所检运动、感觉神经传导速度均未见异常。临床诊断为HSP可能性大。随后对患者DNA进行了目标区域捕获测序和Sanger测序验证,并未发现HSP的基因位点突变,却发现ABCD1基因的exon 2存在c.961_963del(编码区第961-963号核苷酸缺失)的核苷酸变异,该变异导致p.321_321del(第321号氨基酸缺失),为缺失突变。分别在HGMDpro、千人基因组(1000 Genomes)、Exome Aggregation Consortium (ExAC)以及NHLBI Exome Sequencing Project (ESP)数据库查询该突变位点,目前均未见报道 (http://www.hgmd.cf.ac.uk/ac/index.php;http://browser.1000genome s.org/Homo_sapiens/Search/Results?site=ensembl& q=ABCD1;http://exac.broadinstitute.org/gene/ENS G00000101986;http://evs.gs.washington.edu/EVS/ServletManager?variantType=snp&popID=AfricanAm erican&popID=EuropeanAmerican&SNPSummary.x=41&SNPSummary.y=1&SNPSummary=Display+SNP+Summary)。随后进行了家系验证(图1)示,患者母亲存在该基因位点的杂合突变,患者父亲无该基因位点的突变。该基因突变在正常人群中的携带尚未见报道。例1转录本编号NM_000033;测序深度3/73(0.96),覆盖率1.000。修正诊断为AMN。予以巴氯芬降低肌张力、B族维生素营养神经等治疗,出院1年后电话随访,患者症状较前无明显变化。
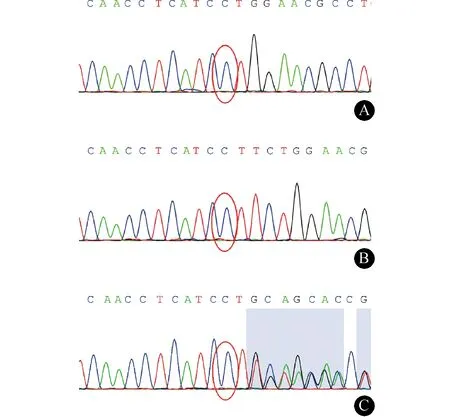
图1 例1患者及家系Sanger测序验证结果。A:例1患者在chrX:152994747-152994749 exon2存在c.961_963del的纯合突变;B:例1患者之父在该位点无突变;C:例1患者之母在该位点存在c.961_963del的杂合突变
1.2 例2 男,50岁,主因“进行性行走不利5年,小便失禁3年”于2014年8月入院。5年前无明显诱因出现双下肢发僵、无力,行走不利,症状逐渐加重。3年前出现小便失禁,性生活不能,无肌肉萎缩、感觉障碍,双上肢活动无异常。外院查头MRI未见明显异常。为求进一步诊治收入我院。病后体质量无减轻。其母亲生前约40~50岁开始出现行走不稳,具体不详。入院查体:神清、语利。高级皮质功能及颅神经检查未见明显异常。双上肢肌力、肌张力、腱反射正常。双下肢肌张力增高,肌力Ⅳ级,腱反射()。深浅感觉查体未见明显异常。双侧Hoffmann征、Babinski征阳性。入院后完善检查:血尿便常规、肝肾功能、肿瘤标志物、叶酸、维生素B12、甲状腺功能均未见异常。CSF常规、生化、免疫、病毒检查均未见明显异常。颈胸髓MRI示颈胸髓较细。EMG示上下肢周围神经源性损害(运动、感觉均受累,髓鞘损害为主)。临床诊断为脊髓周围神经病变,HSP待排除。随后对患者DNA进行了目标区域捕获测序,发现患者ABCD1基因的exon 1存在c.310C>T(编码区第310号氨基酸由C变成T)的核苷酸变异,该变异导致p.R104C[第104号氨基酸由R(精氨酸)变成C(半胱氨酸)],为错义突变。该基因位点曾于1995年报道为致病突变[4],且该基因突变在正常人群中的携带尚未见报道。由于该患者的母亲已去世,因此未行家系验证。例2转录本编号NM_000033;测序深度0/14 (1.00),覆盖率0.999。修正诊断为AMN。予以巴氯芬降低肌张力、B族维生素营养神经等治疗,出院2年后电话随访,患者症状较前无明显变化。
1.3 例3 男,35岁,主因“双下肢无力7年,小便失禁1年”于2015年5月入院。患者于7年前无明显诱因出现双下肢无力,不影响行走。1年前症状较前加重,上楼困难,行走拖拽,伴小便失禁。于当地医院行头颅MRI平扫未见明显异常。EMG示双下肢运动神经传导速度减慢,末端潜伏期延长,感觉神经传导速度未见异常。为求进一步诊治收入我院。病后体质量无明显减轻,其母亲及大姨可疑有双下肢无力的症状。入院查体:神清、语利,高级皮质功能及颅神经查体未见明显异常,双上肢肌力Ⅴ级,双下肢肌力Ⅳ级,四肢肌张力正常。四肢腱反射(),深浅感觉查体未见明显异常。双侧Hoffmann征、Babinski征均阳性。入院后化验:血尿便常规、肝肾功能、甲状腺功能、叶酸、维生素B12等均未见明显异常。头MRI未见脑白质异常信号。头MRS未见异常谱峰。颈胸段MRI示颈胸段脊髓萎缩改变。临床诊断为HSP可能性大。对患者DNA行目标区域捕获测序和Sanger测序验证,发现其ABCD1基因的exon 3存在c.1202G>A(编码区第1202号核苷酸由G变成A)的核苷酸变异突变,该变异导致p.R401Q[第401号氨基酸由R(精氨酸)变成Q(谷氨酰胺)],为错义突变。该基因突变已被报道为致病突变[5],且该基因突变在正常人群中的携带尚未见报道。随后进行了家系验证(图2)示,患者母亲存在该基因位点的的杂合突变。例3转录本编号NM_000033;测序深度4/39(0.91),覆盖率0.999。修正诊断为AMN。予以巴氯芬降低肌张力、B族维生素营养神经等治疗。患者出院后复诊时查血皮质醇示结果正常,极长链脂肪酸(VLCFAs)示结果为阳性[C26:0 1.14 μmol/L (0.30~0.70 μmol/L),C24:0/C22:0 1.65 (0.00~0.94),C26:0/C22:0 0.03 (0.000~0.018)] ;1年后电话随访,患者症状较前无明显变化。
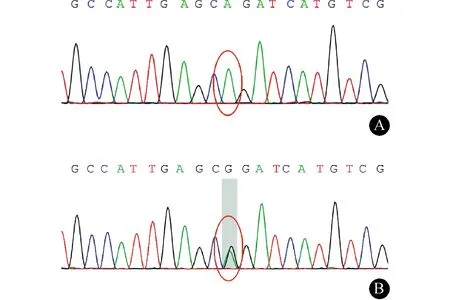
图2 例3患者及家系Sanger测序验证结果。A:例3患者在chrX:153001686 exon3存在c.1202G>A的纯合突变;B:例3之母在该位点存在c.1202G>A的杂合突变
2 讨 论
ALD是最常见的溶酶体病之一,属于X-连锁遗传病,其致病基因为位于X染色体上的ATP结合匣D亚组膜1(ABCD1)。该基因突变导致VLCFAs不能转膜进入细胞溶酶体进行脂肪酸氧化,而在细胞和体液内异常堆积,特别是在脑白质、脊髓、肾上腺及睾丸中,使细胞和血浆中VLCFAs水平升高,出现弥散性神经脱髓鞘和肾上腺皮质功能不足的临床表现[6]。AMN是ALD最常见的一种临床表型,主要累及脊髓,可伴周围神经、脑白质病变,临床上以缓慢进展的痉挛性截瘫为主要特征,可伴有小便障碍、周围神经损害。HSP是以双下肢痉挛性瘫痪为主的神经系统退行性疾病,可呈常染色体显性遗传、常染色体隐性遗传、X染色体连锁遗传、母系遗传,根据临床表型可分为单纯型与复杂型[3]。单纯型主要表现为双下肢痉挛性瘫痪、排尿障碍,双上肢不受累,症状进展缓慢;复杂型可合并有其他神经系统或非神经系统的临床表现,如认知障碍、肌张力障碍、癫痫、小脑性共济失调、周围神经病变、性格内向、情绪不稳定等[3]。
肾上腺脊髓神经病以缓慢进展的双下肢无力、发僵为主要临床表现,可伴有小便障碍,可不伴有肾上腺皮质功能减退的临床特征,可不伴有脑白质异常信号,因此容易被误诊为HSP。不伴有周围神经损害的AMN易误诊为单纯型HSP[7],伴有周围神经损害的AMN易误诊为复杂型HSP。
就疾病遗传方式来讲,AMN呈X连锁隐性遗传,而HSP呈多种遗传方式,包括常染色体隐性遗传、常染色体显性遗传及X连锁隐性遗传[3]。本文中3例病例的疾病遗传史均不明确,例2患者的母亲、例3患者的母亲及大姨有可疑症状,但均不明确。因此对于以痉挛性截瘫为主要临床表现且不伴有脑白质异常信号的患者,尤其是男性患者,需要特别注意与AMN相鉴别。
就临床症状来讲,AMN与HSP很难鉴别。这两种疾病均隐袭起病,缓慢进展,以双下肢痉挛性瘫痪为主要表现,均可伴有小便障碍、周围神经损害等。本文中提到的3例病例,均表现为双下肢痉挛性瘫痪,伴或不伴小便障碍、周围神经损害,完全符合HSP的典型临床特点。30%的AMN病例在出现神经系统病变时可不伴肾上腺功能皮质功能异常[8],AMN患者既可出现肾上腺功能不全的临床表现,也可有临床下的促肾上腺皮质激素增高或正常[9-12]。本文中3例患者均无肾上腺皮质功能异常的临床表现,也无临床下的促肾上腺皮质激素增高或正常。
就影像学表现来讲,HSP患者和AMN患者均可伴有脊髓萎缩[13-14],且相当一部分HSP患者与60%的AMN患者的头MRI检查可不伴有大脑白质异常信号[1,8,15]。本文中3例患者头MRI均未见脑白质异常信号,2例伴有可疑脊髓萎缩。因此影像学方面也难以将HSP与AMN区分开来。EMG检查HSP与AMN均可伴有周围神经的损害,且复杂型HSP与AMN伴有的周围神经损害均多以轴索损害为主[2-3,16],故难以区分二者。
在实验室检测方面,尽管VLCFAs对于诊断ALD具有重要的意义[17]。但也有研究[18-19]报道AMN患者不伴有VLCFAs的增高。这可能与饮食中含有较高的芥酸或实验室检查水平偏低等有关[20]。而且有研究[6,21]显示,15%的ALD女性患者可不伴有VLCFAs增高。因此, VLCFAs正常并不能完全排除ALD。HSP与AMN最终仍要靠基因检测来鉴别。
本文回顾性分析了3例曾被误诊为HSP的ALD的临床资料,总结了HSP与AMN的鉴别要点在于诊断HSP时,要注意与AMN相鉴别,尤其是男性患者,在缺乏典型的疾病遗传史时,需进行VLCFAs的筛查,有条件者行基因检查以确诊。
[1] Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy[J]. Nat Clin Pract Neurol, 2007, 3: 140.
[2] Engelen M, Kemp S, de Visser M, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management[J]. Orphanet J Rare Dis, 2012, 7: 51.
[3] Finsterer J, Löscher W, Quasthoff S, et al. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance[J]. J Neurol Sci, 2012, 318: 1.
[4] Ligtenberg MJ, Kemp S, Sarde CO, et al. Spectrum of mutations in the gene encoding the adrenoleukodystrophy protein[J]. Am J Hum Genet, 1995, 56: 44.
[5] Fuchs S, Sarde CO, Wedemann H, et al. Missense mutations are frequent in the gene for X-chromosomal adrenoleukodystrophy (ALD)[J]. Hum Mol Genet, 1994, 3: 1903.
[6] Schackmann MJ, Ofman R, van Geel BM, et al. Pathogenicity of novel ABCD1 variants: The need for biochemical testing in the era of advanced genetics[J]. Mol Genet Metab, 2016, 118: 123.
[7] Shaw-Smith CJ, Lewis SJ, Reid E. X-linked adrenoleukodystrophy presenting as autosomal dominant pure hereditary spastic paraparesis[J]. J Neurol Neurosurg Psychiatry, 2004, 75: 686.
[8] 何玺玉. X-连锁肾上腺脑白质营养不良的诊断与治疗[J].中华实用儿科临床杂志, 2015, 30: 561.
[9] 毛晨晖,谢曼青,刘彩燕, 等.表现为痉挛性截瘫的成人型脑白质营养不良[J].中华神经科杂志, 2015, 48: 748.
[10] Park HD, Park SJ, Choi YM, et al. Adrenomyeloneuropathy presenting with adrenal insufficiency[J]. Ann Rehabil Med, 2013, 37: 563.
[11] Dubey P, Raymond GV, Moser AB, et al. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening[J]. J Pediatr, 2005, 146: 528.
[12] Kemp S, Huffnagel IC, Linthorst GE, et al. Adrenoleukodystrophy- neuroendocrine pathogenesis and redefinition of natural history[J]. Nat Rev Endocrinol, 2016, 12: 606.
[13] Hedera P, DiMauro S, Bonilla E, et al. Phenotypic analysis of autosomal dominant hereditary spastic paraplegia linked to chromosome 8q[J]. Neurology, 1999, 53: 44.
[14] Suryawanshi A, Middleton T, Ganda K. An unusual presentation of X-linked adrenoleukodystrophy[J]. Endocrinol Diabetes Metab Case Rep, 2015, 2015: 150098.
[15] Kumar AJ,Köhler W,Kruse B,et al.MRI findings in adult-onset adrenoleukodystrophy[J].AJNR Am J Neuroradiol,1995,16:1227.
[16] Engelen M, Barbier M, Dijkstra IM, et al. X-linked adrenoleukodystrophy in women: a cross-sectional cohort study[J]. Brain, 2014, 137: 693.
[17] Wiesinger C, Eichler FS, Berger J. The genetic landscape of X-linked adrenoleukodystrophy: inheritance, mutations, modifier genes, and diagnosis[J]. Appl Clin Genet, 2015, 8: 109.
[18] Koutsis G, Lynch DS, Tucci A, et al. A novel ABCD1 mutation detected by next generation sequencing in presumed hereditary spastic paraplegia: A 30-year diagnostic delay caused by misleading biochemical findings[J]. J Neurol Sci, 2015, 355: 199.
[19] Karimzadeh P, Jafari N, Nejad-Biglari H, et al. The clinical features and diagnosis of adrenoleukodystrophy: a case series of iranian family[J]. Iran J Child Neurol, 2016, 10: 61.
[20] Moser AB, Kreiter N, Bezman L, et al. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls[J]. Ann Neurol, 1999, 45: 100.
[21] O’Neill BP, Moser HW, Saxena KM, et al. Adrenoleukodystrophy:clinical and biochemical manifestations in carriers[J]. Neurology, 1984, 34: 798.
Analysisofmisdiagnosisofadrenalleukodystrophycharacterizedwithspasmodicparaplegiabutwithoutabnormalcerebralwhitemattersignals(reportof3cases)
LUYan,LIUYan-qiu,ZHANGXin-qing,etal.
DepartmentofNeurology,XuanwuHospitalofCapitalMedicalUniversity,Beijing100053,China
ObjectiveTo ovserve the clinical features of adrenal leukodystrophy (ALD) characterized with spasmodic paraplegia but without abnormal cerebral white matter signals, and to avoid to be diagnosed as hereditary spastic paraplegia (HSP).MethodsThe clinical data of 3 ALD patients who were main characterized as spasmodic paraplegia and without abnormal cerebral white matter signals and misdiagnosed as HSP were reviewed retrospectively.ResultsThree patients were all manifested as significant spastic paraplegia with an insidious onset and slow progression, and with or without autonomic nerve or peripheral nerve impairment. There were no abnormal signals in the brain white matter. Three patients were all diagnosed as adrenomyeloneuropathy (AMN) by next generation sequencing after the misdiagnosis as HSP. Gene sequencing showed that ABCD1 gene exon 2 c.961_963del deletion mutantion, ABCD1 gene exon 1 c.310C>T missense mutation and ABCD1 gene exon 3 c.1202G>A missense mutation, separately. The family verification of case 1 and case 2 showed that both of their mothers had corresponding genetic mutations.ConclusionsAMN is one clinical manifestation of ALD, characterized as spastic paraplegia in the lower limbs. Patients without abnormal siginals in brain white matter are easily to be misdiagnosed as HSP. So plasma very long chain fatty acids (VLCFA) testing or gene testing should be used for the patients who are clinically suspected HSP, especially in male patients, in order to identify ALD.
adrenal leukodystrophy;adrenomyeloneuropathy;hereditary spastic paraplegia;gene testing;differential diagnosis
R742.8
A
1004-1648(2017)05-0373-04
100053北京,首都医科大学宣武医院神经内科(卢岩和刘艳秋是共同第一作者)
张新卿
2016-12-29
2017-01-31)
猜你喜欢
中国现代医生(2022年21期)2022-08-22
基层中医药(2022年1期)2022-07-22
全科护理(2022年3期)2022-02-18
云南中医中药杂志(2020年3期)2020-05-11
中成药(2019年12期)2020-01-04
中国医药科学(2017年9期)2017-08-04
中国现代神经疾病杂志(2017年1期)2017-03-29
放射学实践(2016年6期)2016-12-15
广东药科大学学报(2016年6期)2016-03-10
西南医科大学学报(2016年4期)2016-01-03
