
辣椒素固体分散体的处方工艺优化及表征Δ
2019-06-25 01:56游国叶樊轻亚杜晶李会娜信阳职业技术学院药学院河南信阳464000杭州民生药业有限公司杭州30000
中国药房 2019年11期
游国叶,樊轻亚,杜晶,李会娜(.信阳职业技术学院药学院,河南信阳464000;2.杭州民生药业有限公司,杭州 30000)
辣椒(Capsicum frutescensL.)属茄科辣椒属,又名海椒、番椒、辣茄等,为植物辣椒的干燥成熟果实,其温中散寒,开胃消食,用于治疗寒滞腹痛、呕吐、泻痢、冻疮等多种病症[1]。辣椒素(Capsaicin,CAP)是从辣椒属植物中得到的药用活性成分,分子式为C18H27NO3,是一种无色无味的生物碱,能溶于乙醇、乙醚、苯及氯仿,微溶于二硫化碳[2]。现代药理学研究证明,CAP具有止痒镇痛[2-3]、减肥调脂[2]、抗癌[2,4]、抗炎和抗疲劳[2,5]、降血压[6]、调节内分泌系统[4]、保护心脑血管和消化系统[7]等功效,可用于治疗神经疼痛、关节炎、肥胖、糖尿病以及癌症等疾病。CAP依据生物药剂学分类系统属于Ⅳ类[8],溶解度小、渗透性低。CAP难溶于水、刺激性大、生物利用度低,限制了其医药用途。
固体分散体(Solid dispersion,SD)概念于1964年被Sekiguchi K等[9]提出,是利用一定的技术使药物以固体溶液、微晶或无定型状态高度分散在载体中所形成的分散体系。SD中药物粒子因粒径减小,表面自由能显著增大,从而提高了难溶性药物的溶解度、溶出度和生物利用度。目前固体分散技术已广泛用于增加难溶性药物的溶解度和溶出速率,提高溶出度是改善难溶性药物生物利用度最有效的措施。有研究报道,将灯盏花素、沙棘总黄酮等难溶性药物制备成SD均显著提高了药物的溶出度[10-11]。SD可以是最终产品,也可作为剂型中间体[12-13],这对于研究CAP的产品化和后开发具有重要意义。
在SD制备中,表面活性剂类的载体能更有效地增加药物溶出度,同时阻碍药物重结晶,防止SD老化。本研究采用固体分散技术,以泊洛沙姆188(P188)或聚乙二醇4000(PEG4000)为载体,制备了CAP-SD,以体外累积释放度为考察指标,在前期单因素考察基础上,采用正交试验优化处方,并通过差式扫描量热法(DSC)和X射线粉末衍射法(XRD)探究CAP在SD中的分布形态,为CAP制剂的开发提供参考。
1 材料
1.1 仪器
TG328A型分析天平(上海精科仪器有限公司);TD-3500型XRD仪(丹东通达科技有限公司);Quanta 400型扫描电镜(美国FEI公司);RC-6型智能溶出仪(上海巴玖实业有限公司);LC-10AT型高效液相色谱仪,包括SPD-10AVP型紫外检测器、CBM-102型色谱工作站(日本岛津公司);TU-1810型紫外-可见分光光度计(北京普析通用仪器有限责任公司);KQ5200DB型数控型超声波清洗器(昆山超声波仪器有限公司);DHG-9140A型电热恒温鼓风干燥箱和DZF-6210型真空干燥箱(上海沪粤明科学仪器有限公司);Diamond DSC型DSC分析仪(美国Perkin-Elmer公司);LHH-150SD型药品稳定性试验箱(上海一恒科学仪器有限公司);DJ-1型磁力搅拌器(江苏省金坛市佳美仪器有限公司)。
1.2 药品与试剂
辣椒油(天津圣惠生物科技有限公司,批号:20160928-015,纯度:以CAP计7.06%);CAP对照品(中国食品药品检定研究院,批号:100829-201605,纯度:95%);P188和PEG4000(国药集团化学试剂有限公司,批号:20160203011、HTF20160112-006);甲醇为色谱纯,其余试剂均为分析纯。
2 方法与结果
2.1 CAP浓缩物的制备
取辣椒油适量,加乙醇20 mL超声(频率:40 kHz,功率:200 W,每10 min停止,振摇后继续,下文中其他超声操作无特殊说明者均以此条件进行)30 min,过滤,收集滤液备用;滤渣中加甲醇20 mL继续超声30 min后过滤,收集滤液,重复操作1次,合并3次的滤液,70℃恒温干燥箱内浓缩,浓缩成流浸膏,CAP含量为40%(g/mL)。
2.2 CAP-SD的制备
采用熔融法制备CAP-SD,分别称取100 mL的CAP浓缩物、200 g P188和120 g PEG4000),先将P188和PEG4000在65℃水浴中不断搅拌至完全熔化制成熔融态载体,再加入CAP浓缩物继续搅拌适宜时间,然后迅速转移至冰浴中冷却至完全固化,最后放置于真空干燥箱中干燥24 h,取出,研磨粉碎过60目筛得CAP-SD。
2.3 物理混合物的制备
分别称取处方量的CAP浓缩物及辅料(P188、PEG4000),3次过筛混合均匀,即得物理混合物。
2.4 CAP含量测定方法的建立
2.4.1 溶液 ①CAP对照品溶液。精密称取CAP对照品50 mg,置于100 mL量瓶中,加入无水乙醇超声溶解,定容,摇匀;准确量取1.0 mL,置于10 mL量瓶中,加流动相定容,摇匀,稀释制成质量浓度为50 μg/mL的对照品溶液。②供试品溶液。精密称取CAP-SD适量(约相当于CAP 50 mg),置于100 mL量瓶中,加入无水乙醇超声溶解,定容,摇匀,准确量取1.0 mL,置于10 mL量瓶中,加流动相定容,摇匀,稀释制成质量浓度为50 μg/mL供试品溶液。③阴性对照品溶液。因CAP在水中溶解度极小,如体系中加入适当比例的十二烷基硫酸钠(SLS-Na)后,可增加其溶解度,再则笔者前期研究发现将CAP制备成CAP-SD后采用脱气处理过的含3%SLS-Na的纯水作为释放介质进行体外释放度试验,满足漏槽条件。故阴性对照品溶液制备时,取处方量的辅料(P188、PEG4000)及适当比例量的SLS-Na混合物,不含CAP,用流动相溶解、稀释,定容,制成阴性对照品溶液。
2.4.2 检测波长 依据2015年版《中国药典》要求及课题组前期研究成果[14]和相关文献报道[15],取CAP对照品溶液和阴性对照品溶液适量,用流动相定容后进行紫外扫描。结果显示,CAP在280 nm波长处有最大吸收,且阴性对照品在此处无干扰,故选择280 nm作为CAP的检测波长。紫外扫描图谱见图1。
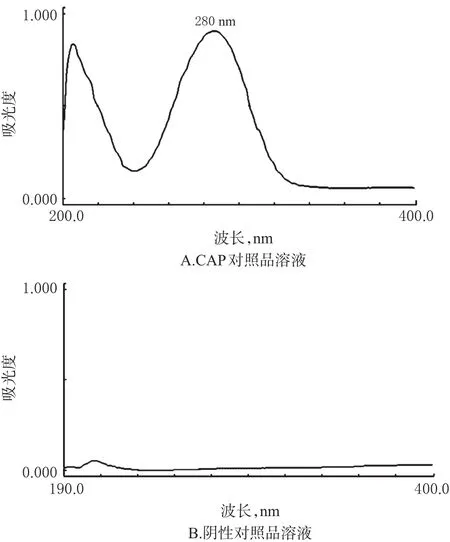
图1 紫外扫描图谱Fig 1 UV scanning spectrum
2.4.3色谱条件与专属性色谱柱:Kromasil-C18(250 mm×4.6 mm,5 μm);流动相:甲醇-水(80∶20,V/V);检测波长:280 nm;柱温:30 ℃;流速:1.0 mL/min;进样量:20 μL。取CAP对照品溶液、供试品溶液、阴性对照品溶液、阴性对照品+CAP对照品的混合液分别进样测定,记录色谱。结果显示,CAP的保留时间为6.5 min,且峰形较好,无前沿、拖尾现象,与相邻色谱峰的分离度均大于1.5,其他成分对CAP的测定无干扰,理论板数按CAP峰计不低于3 000。色谱图见图2。
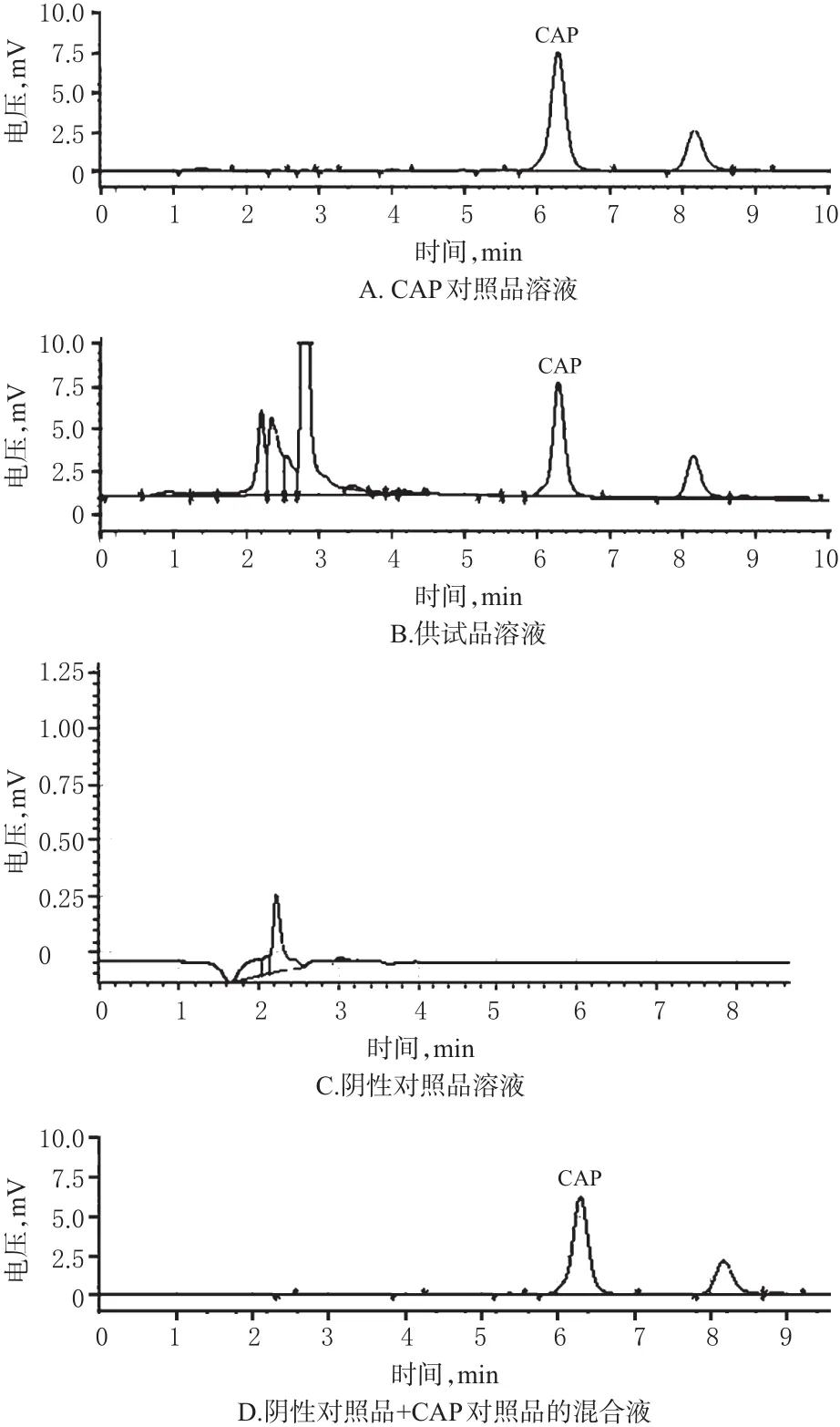
图2 高效液相色谱图Fig 2 HPLC chromatograms
2.4.4 线性关系、检测限、定量限 精密称取CAP对照品50 mg,置于100 mL量瓶中,加入无水乙醇超声溶解,定容,制成500 μg/mL CAP对照品贮备液。精密量取该贮备液0.2、1、2、3、5、7、9 mL,分别置于10 mL量瓶中,加流动相定容,按照“2.4.3”项下色谱条件进样测定,每个浓度进样3次,记录峰面积。以峰面积(A)为纵坐标,CAP质量浓度(c)为横坐标,进行线性回归。得回归方程为A=2 903.9c+91 279(r=0.999 3),CAP检测质量浓度的线性范围为10~450 μg/mL。在该色谱条件下,以信噪比≥3∶1和信噪比≥10∶1分别确定检测限和定量限,结果显示,检测限为0.02 μg/mL,定量限为0.1 μg/mL。
2.4.5 精密度 精密量取CAP对照品溶液适量,以流动相稀释制成质量浓度分别为50、150、350 μg/mL的CAP溶液3份,按照“2.4.3”项下色谱条件进样,每份样品隔4 h测定1次,共测定6次,考察日内精密度;每份样品每日测定1次,连续测定6 d,考察日间精密度。结果显示,日内、日间RSD分别为0.98%(n=6)、1.02%(n=6),表明精密度良好。
2.4.6 重复性 取同一批次CAP-SD(批号:20170525)6份,按照“2.4.1②”项下方法制成供试品溶液,再按照“2.4.3”项下色谱条件进样测定,分析每份供试品溶液中CAP含量。结果显示,含量的RSD为0.79%(n=6),表明重复性良好。
2.4.7 稳定性 取供试品溶液适量,分别放置0、3、6、9、12、18、24 h后,按照“2.4.3”项下色谱条件进样测定,分析供试品溶液中CAP含量。结果显示,含量的RSD为0.99%(n=7),表明供试品溶液在24 h内稳定。
2.4.8 准确度 取已知CAP含量的CAP-SD(批号:20170525)适量,分别加入低、中、高质量浓度(150、350、450 μg/mL)CAP对照品溶液,各3份,按照“2.4.3”项下色谱条件进样测定,分析CAP含量,以测得量/真实量计算回收率,结果见表1。
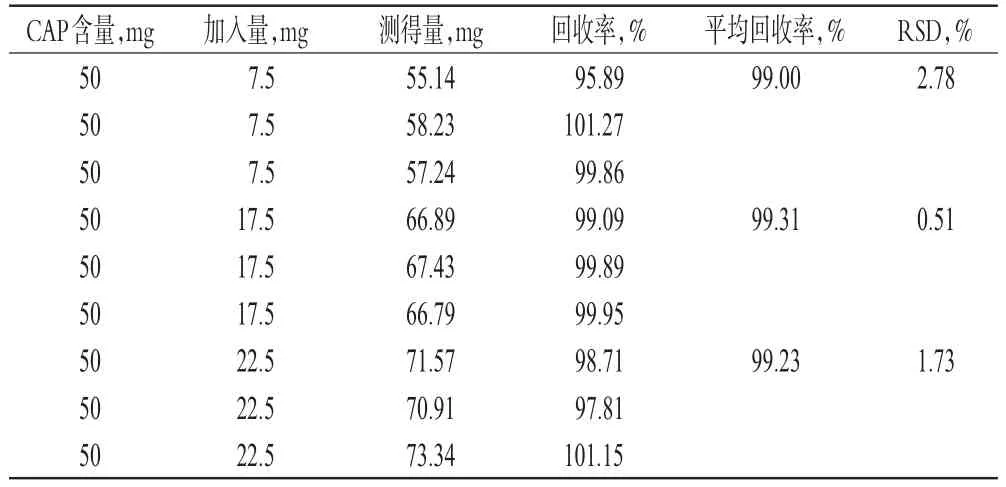
表1 回收率测定结果Tab 1 Results of recovery tests
2.4.9 CAP含量 精密称取CAP-SD(批号:20170525)适量(约相当于CAP 50 mg),加入无水乙醇超声溶解,以流动相定容至刻度,摇匀,过滤,按照“2.4.3”项下色谱条件进样测定,记录峰面积,代入回归方程计算CAP含量,结果显示,CAP的测得量为49.94 mg(n=3),即CAP-SP中CAP的含量为10.89%。
2.5 体外释放度的测定
按照2015年版《中国药典》四部0931溶出度及释放度测定法第二法(桨法)测定[16],设定转速为(50±1)r/min、水浴温度为(37±0.5)℃,释放介质为脱气处理过的含3%SLS-Na的纯水900 mL。称取CAP和含等量CAP的CAP-SD约500 mg,置于溶出杯中,自药物接触释放介质开始计时,分别于5、15、30、45、60、120、180、240、300、480 min取样5 mL,并及时补加5 mL 等温释放介质,取样液经0.45 μm微孔滤膜滤过,吸取续滤液,置于10 mL量瓶中,按照“2.4.3”项下色谱条件进样测定,计算CAP的累积释放度。
2.6 处方优化
根据笔者前期单因素考察结果发现,载体种类、药载比及熔融态载体与药物搅拌时间对CAP-SD的累积释放度有显著影响,故本试验以载体种类(A)、药载比(B)、搅拌时间(C)为考察因素,以60 min累积释放度为评价指标,每个因素3水平,设计正交试验优化处方。因素与水平见表2,正交试验设计与结果见表3,方差分析结果见表4。

表2 因素与水平Tab 2 Factors and levels
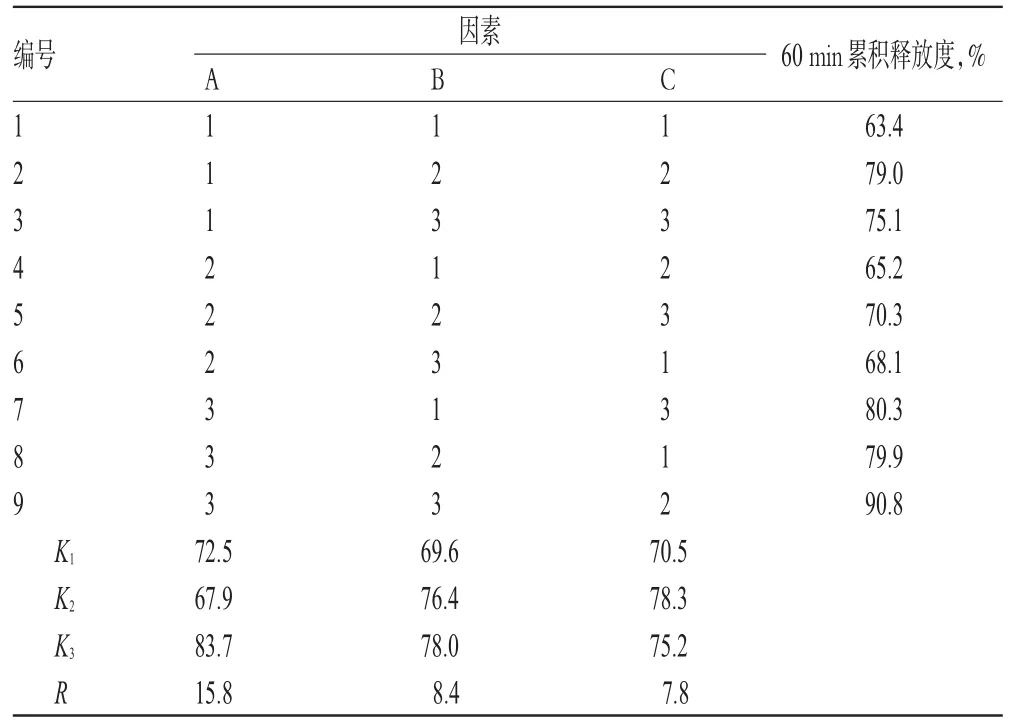
表3 正交试验设计与结果Tab 3 Design and results of orthogonal test
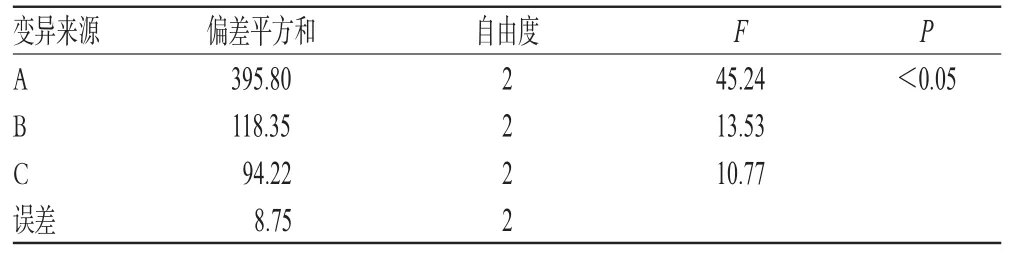
表4 方差分析结果Tab 4 Results of variance analysis
由表3结果显示,3个因素对CAP-SD体外释放度的影响顺序依次为A>B>C。由表4结果显示,A因素对CAP-SD的60 min累积释放度有显著影响(P<0.05),综合考虑,CAP-SD最优处方为A3B3C2,即CAP-SD处方中载体为P188和PEG4000,CAP-P188-PEG4000的质量比为1∶5∶3,采用熔融态载体与药物搅拌时间为20 min。按最优处方制备3批CAP-SD样品,考察其60 min累积释放度,并采用相似因子(f2)法判断3批样品释放曲线的差异。结果显示,3批样品的释放曲线两两之间的f2值均大于50,不存在显著差异,其60 min累积释放度分别为87.8%、85.7%、80.2%(n=3),因此可判定本处方工艺重现性良好。
2.7 稳定性试验
按“2.6”项下最优处方制备CAP-SD,批号为20170806,置于LHH-150SD型药品稳定性试验箱(温度为40℃、相对湿度为75%),分别放置于0、30、180 d取样,通过外观色泽、含量及XRD判断其在考察期内的稳定性。结果显示,CAP-SD放置0、30、180 d后外观色泽无变化;CAP含量依次为10.98%、11.01%、10.95%,RSD为3%(n=3);XRD结果显示未出现新峰,提示CAP-SD具有良好的物理稳定性。稳定性试验XRD曲线见图3。
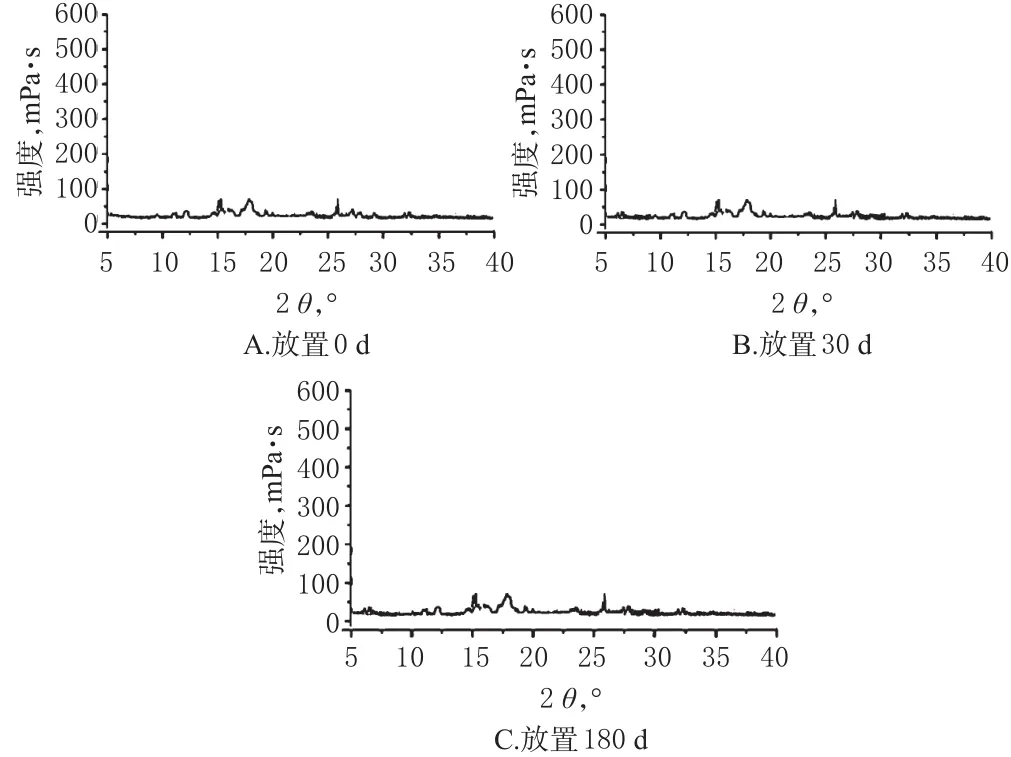
图3 稳定性试验XRD曲线Fig 3 DSC curves of stabilitytests
2.8 CAP-SD的物相验证
2.8.1 DSC 以空白铝坩埚为参比,N2为保护气,温度为20~200℃,升温速度为10℃/min,分别对CAP对照品、P188、PEG4000、物理混合物及CAP-SD进行DSC分析。DSC曲线见图4。
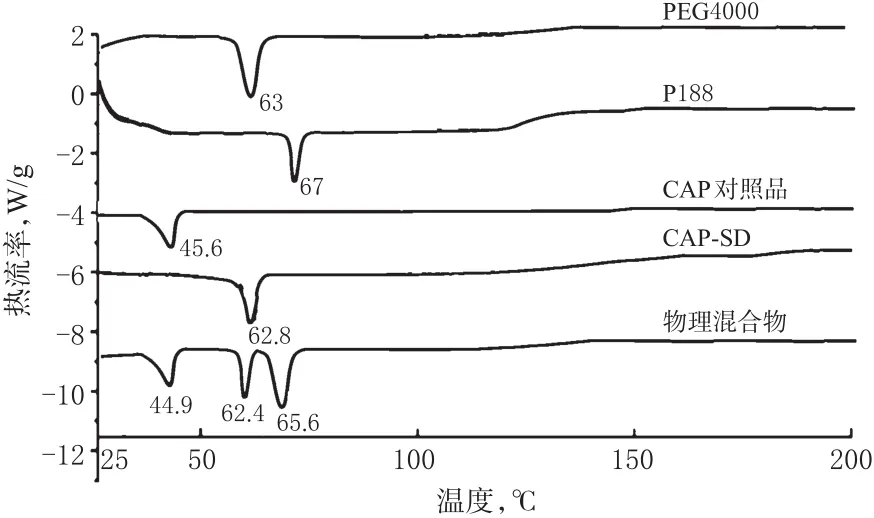
图4DSC曲线Fig 4 DSC curves
由图4显示,CAP对照品中有1个熔融峰,在45.6℃;载体材料PEG4000和P188各自有1个熔融峰,分别在63℃和67℃,且曲线起伏度大;物理混合物有3个吸热峰,分别在44.9、62.4、65.6℃,依次为CAP峰和两辅料峰,由此可见,物理混合物中CAP与辅料之间没有发生相互作用,药物仍以晶体状态存在;CAP-SD有1个吸热峰,在62.8℃,CAP吸热峰消失,该峰与载体P188和PEG4000峰相比均前移,说明CAP与载体P188、PEG4000形成低共熔物或共沉淀物,即CAP以分子形式或者无定型状态存在。
2.8.2 XRD 测试条件为Cu-Ka靶,电压为40 kV,电流为30 mA,扫描速率为10°/min,步长为0.02°,扫描范围为5°~40°,发散狭缝与防散射狭缝均为1°。分别对CAP对照品、处方量P188和PEG4000的混合物、物理混合物及CAP-SD进行XRD分析。XRD曲线见图5。
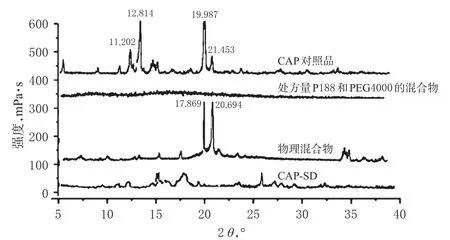
图5XRD曲线Fig 5 XRD curves
由图5显示,CAP在11.202°、12.814°、19.987°、21.453°有数个强衍射峰,证明CAP具有晶体结构;P188和PEG4000的混合物在监测范围内未检测到特征衍射峰;物理混合物在17.869°、20.694°有两个强衍射峰;CAP-SD中药物的结晶衍射峰消失或极大减弱,表明CAP在形成SD后,载体对CAP有抑晶作用,使CAP以无定型状态或分子状态高度分散于载体中。
3 讨论
CAP具有广泛的药理活性及食疗作用,但由于溶解度小、口感辛辣,临床应用受限,故而加强CAP应用的关键在于提高其溶解度、增加其生物利用度、改善其口感。SD利用亲水性的高分子材料及材料本身具备的抑晶、高度分散作用,将难溶性药物以分子、离子状态分散于载体材料中,极大地提高了药物的溶解度,并且能有效地掩盖药物自身的辛辣刺激感。在SD载体材料选择上,除了考虑其安全性、稳定性,还需具备一定的理化性质才适用于SD的制备,这些理化性质包括玻璃态化转化温度(Tg)、溶解度参数、熔融黏度、载体材料与药物之间的相互作用等。CAP属于脂溶性物质,其表观溶解度为(22.85±0.06)mg/L[17],极微溶于水,SD制备过程中使用半极性物质PEG4000为载体,可借助PEG4000较强的润湿性和分散性,使得极性较差的CAP润湿性能增强,即亲水能力增加,与载体材料接触面积亦增大,在与溶出液接触后可加快药物溶出;另外P188是一种由聚氧乙烯(PEO)、聚氧丙烯(PPO)组成的PEO-PPO-PEO非离子型三嵌段共聚物,具有独特的疏水内核-亲水外壳结构,能与许多药物形成孔隙固溶体,使CAP高度分散于P188中,再结合PEG4000的润湿增溶能力,进一步增加了CAP的溶出,改善其吸收,而且聚合物胶束增溶作用和药物与聚合物分子间的作用相互反应,使得药物溶解度和溶出速率增加,生物利用度也得以提高。此外,PEG4000和P188都是亲水性聚合物,彼此分子之间以及与药物分子之间均可能会形成氢键,使得体系更加稳定。本研究以体外累积释放度为考察指标,以载体种类、药载比及搅拌时间为考察因素,通过正交试验优化处方,最终确定最优处方工艺,按最优处方工艺所制的CAP-SD中CAP的体外60 min的累积释放度达到80%,并结合DSC、XRD分析显示,CAP与PEG4000、P188间相容性良好,三者熔点接近且均较低,可快速熔融并迅速冷却,工艺条件简便可控。在CAP-SD中,CAP与PEG4000、P188形成了低共熔物,且CAP以无定型或分子状态状态存在,在初步稳定性考察期内累积释放度与外观均无明显变化,可见采用固体分散技术不仅可解决CAP溶解度低的缺陷,还可以将其制备成质量稳定可靠的中间体,为CAP进一步剂型研究开发提供重要依据。
由于CAP对胃有刺激性,后期笔者欲将CAP-SD制备成肠溶胶囊,所以考察了多种释放介质[含1%~5%SLS-Na纯水溶液及前2 h 0.1 mol/L盐酸溶液之后使用pH 7.4磷酸盐缓冲液(模拟胃肠道溶液)] 、转速、介质温度等,最终结果显示,采用900 mL 3%SLS-Na纯水溶液和模拟胃肠道溶液为释放介质,转速为(50±1)r/min、水浴温度为(37±0.5)℃时,二种释放介质间释放结果无明显差异;且本文主要研究CAP-SD,所以在释放度考察中选择3%SLS-Na纯水溶液作为释放介质模拟肠溶胶囊壳溶解后的释放过程。今后将进一步探究CAP-SD肠溶胶囊在动物体内的吸收状况及其进一步制成制剂的可行性,为天然产物CAP的研究、开发及应用提供指导及参考。
综上所述,本文采用熔融法,以P188和PEG4000制备得到的CAP-SD稳定性良好,药物以无定型状态或分子状态高度分散于载体中,60 min的累积释放度可达80%以上,且工艺条件简便可控,便于规模化生产。但由于试验条件的限制,课题组只进行了初步稳定性考察,所得SD在考察期内稳定,下一步仍需完善其对高温、高湿度、强光照射等因素的稳定性研究。
猜你喜欢
世界科学技术-中医药现代化(2021年5期)2021-11-05
文苑(2020年6期)2020-06-22
中国报道(2018年2期)2018-04-20
试题与研究·中考化学(2016年4期)2017-03-28
中学化学(2016年10期)2017-01-07
中学化学(2015年5期)2015-07-13
小说月刊(2015年1期)2015-04-19
祝您健康(2000年7期)2000-12-29
祝您健康(2000年5期)2000-12-29
