
玉米秸秆消化率全基因组关联分析
2020-12-31 05:58张秋芝史利玉王红武潘金豹
作物杂志 2020年6期
林 淼 张秋芝 史利玉 刘 蓓 王红武 潘金豹
(1北京农学院,102206,北京;2中玉金标记(北京)生物技术股份有限公司,102206,北京)
青贮玉米是草食家畜特别是奶牛最重要的高能量饲料来源。优质青贮玉米具有较高的淀粉含量、低纤维含量和高消化率等特征。玉米秸秆占全株玉米生物量的一半,因此秸秆消化率的高低是影响青贮玉米消化率关键。粗饲料体外干物质消化率(in vitro dry matter digestibility,IVDMD)测定指标有30h和48h两种,48h体外干物质消化率(48h IVDMD)是青贮饲料在草食家畜的消化系统中消化并吸收48h的最大量与进食量的比值。IVDMD体现青贮玉米秸秆利用和转化的效率,主要受遗传因素影响,48h IVDMD是青贮玉米品质育种首要指标[1-4]。
目前,国内外利用不同遗传背景的群体,采用连锁分析的方法,在1~10染色体上均发现了控制玉米秸秆IVDMD的QTL[5-8]。全基因组关联分析(genome-wide association study,GWAS)具有高分辨率且省时等特点,是检测复杂性状变异强大的遗传方法,特别是玉米作为异交作物,重组较多,导致较短的连锁不平衡(linkage disequilibrium,LD),且表型和遗传多样性丰富,分辨率较高[9-11],但不同群体LD存在差异,如农家种1kb、广泛变异群体1.5kb、优良自交系100kb,都利于关联定位的精度[10]。目前关于青贮玉米秸秆消化率性状的全基因组关联分析的研究极少。Wang等[9]以559 285个高质量SNPs对368份玉米自交系采用GWAS方法鉴定了82个秸秆IVDMD显著相关的位点,并发现了直接参与细胞壁生物合成的基因ZmC3H2。但Wang等[9]利用关联分析对秸秆IVDMD进行研究时,群体中没有青贮玉米品种的亲本自交系,从而不利于挖掘更多且直接的优良的青贮玉米消化率性状遗传位点。
本研究以341份玉米自交系为材料,构建关联作图群体,基于全基因组测序的高质量SNPs进行秸秆48h IVDMD的全基因组关联分析,挖掘优良等位变异,筛选候选基因,为优质青贮玉米选育提供参考。
1 材料与方法
1.1 供试材料及田间设计
供试材料为194份美国玉米自交系和147份国内玉米自交系(包括14份国内常用玉米自交系和133份课题组自选玉米自交系)构成的341份关联群体,其中6份为青贮玉米品种的亲本自交系。
2018年5月初将供试材料分别种植于辽宁省沈阳市东亚国际种业公司和内蒙古自治区通辽市农业科学研究院科学研究基地。行长5.0m,行距0.6m,株距0.25m,双行区,自然散粉,田间管理同一般玉米自交系试验田。
1.2 品质测定方法
调查供试材料生育时期(主要包括抽雄期和吐丝期)。在玉米籽粒乳线位置1/2时,选取长势一致的10株玉米,从地上部20cm处刈割,去掉果穗,收获秸秆。用风送式动力铡草机粉碎后混合均匀,平行取两份1 000g装入布袋,立刻放入105℃烘箱中杀青2h,散热散水气后调至65℃烘干至恒重,用锤片式粉碎机磨碎,过40目筛,装入牛皮纸袋,于65℃烘箱烘至恒重,取出样品冷却至室温,待用。
试验采用FOSS近红外光谱仪(NIRS DS2500)[12]对供试样品48h IVDMD(%)进行测定,该仪器采用前分光单色仪技术,探测器是硅(400~1 100nm)和硫化铅(1 100~2 500nm),光谱分辨率是0.5nm-1。将每份样品混匀,取适量于直径为50mm的石英柱形样品杯中,波长为400~2 500nm近红外光以漫反射形式从样杯底部扫描,每份样品扫描2次,每次重复重新取样品,结果取均值(所测定含量是干基)。
1.3 表型统计方法
采用SPSS 20.0进行描述性统计分析和相关性分析,利用R 3.5.1软件对表型频率分布作图,利用Microsoft Excel 2010进行双因素方差分析估算广义遗传力(HB2),HB2=VG/VP×100%=VG(/VG+VE)×100%,VP、VG和VE分别是表型变异方差、遗传变异方差和环境变异方差。
1.4 基因型测定及分析
采用磁珠法[13]提取341份供试材料的基因组DNA,利用Qubit荧光光度计、NanoDrop 2000和1%琼脂糖凝胶电泳分别对样品DNA进行浓度、纯度和完整性检测。使用Illumina PE150测序仪对合格样品DNA进行全基因组重测序,平均测序深度是7×,共得到112 626 032个SNPs标记。采用BWA软件[14]将测序后短序列和玉米B73参考基因组组装V4比对,平均覆盖度是83%,均匀覆盖参考基因组。从112 626 032个SNPs标记中去掉缺失率大于30%、杂合率大于10%、最小等位基因频率小于0.05的标记,剩余6 276 612个高质量SNPs用于全基因组关联分析。
1.5 群体结构和亲缘关系
采用fastStructure 1.0软件[15],以6 276 612个MAF>0.05且均匀分布染色体的SNPs,对341份供试材料进行贝叶斯聚类,计算不同层次的亚群数(K=2~10),再根据最大似然数LnP(D),确定本群体理论上由5大亚群组成。
1.6 秸秆48h IVDMD全基因组关联分析
在考虑群体结构和亲缘关系的情况下,运用MVP软件的FarmCPU模型[16]将秸秆48h IVDMD表型和本群体的基因型进行全基因组关联。Bonferroni校正法即0.05/n(n是本研究的标记数目)确定阈值(P<1.0×10-8),由于此种方法稍严格,故通过文献采用阈值(P<1.0×10-6)[17]。
1.7 秸秆48h IVDMD定位结果比较分析
借助前人用不同群体构建遗传连锁图谱进行的QTL定位结果,若本试验关联定位结果落入前人QTL定位区间内或相差200kb以内说明本试验关联定位效果良好。
1.8 秸秆48h IVDMD候选基因预测
采用Haploview 4.1 jar[18]分析本群体连锁不平衡(LD)衰减距离,本群体LD衰减到0.1时的距离约是2.7kb,在此情况下,为了防止盲目基因或区段扩展,故基于玉米B73基因组组装V4序列信息和GWAS检测到的所有显著SNPs位点(P<1.0×10-6),选取基因内部及其上下游1kb潜在调控区域作为候选基因区段[19-20]。
1.9 群体变异位点分析
利用GATK软件[21]采用Haplotype Caller模式进行变异位点检测,借助ANNOVAR软件[17]对基于候选基因的SNPs进行注释。
2 结果与分析
2.1 秸秆48h IVDMD表型分析
沈阳和通辽两个环境下秸秆48h IVDMD频率分布图和基本描述统计见图1和表1,结果说明遗传变异范围非常广泛,均值分别为80.96%和67.94%,变异系数分别为4.85%和9.50%,说明同一环境的不同自交系间的秸秆48h IVDMD存在差异,不同环境下群体的整体表现也有差异,但整体符合正态分布。Person相关性结果表明,两个环境下秸秆48h IVDMD显著相关。秸秆48h IVDMD的广义遗传力为71.98%。这些结果都说明秸秆48h IVDMD性状主要受到遗传效应影响。
2.2 秸秆48h IVDMD全基因组关联分析
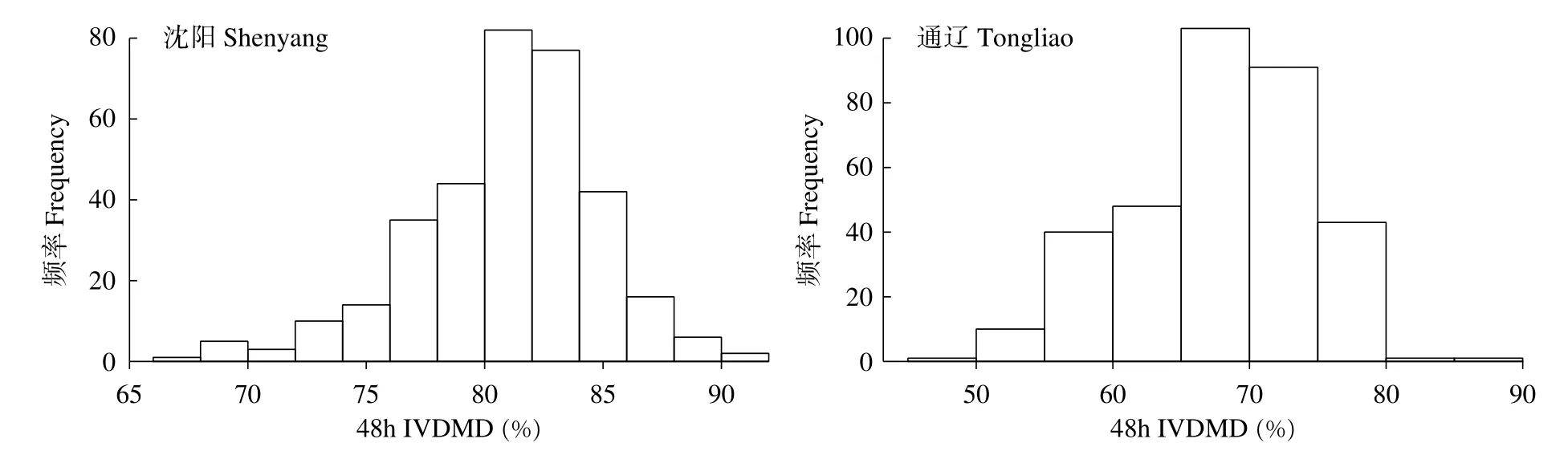
图1 秸秆48h IVDMD频率分布Fig.1 Frequency distribution of 48h IVDMD
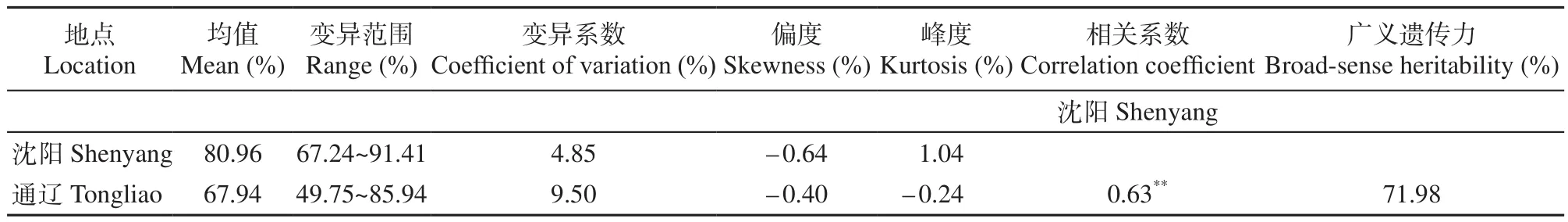
表1 两个环境秸秆48h IVDMD统计分析Table 1 Statistics analysis of 48h IVDMD in two environments
利用FarmCPU模型对341份玉米自交系的48h IVDMD进行全基因组关联分析。绘制曼哈顿图(Manhattan)(图2),超过水平线(两条水平线代表全基因组显著水平阈值6.0和8.0)的SNPs即为与本试验目标性状显著相关的位点。两个环境中FarmCPU模型的QQ图表示群体结构较为良好地被控制(图3)。在沈阳和通辽的两个环境中共检测到153个显著SNPs位点(P<1.0×10-6),分布在玉米第1~10染色体,其中,沈阳环境中,在显著水平P<1.0×10-8上检测到4个显著SNPs位点,分布于第2、3、6、8染色体上(图4)。在第1~3、6~8、10染色体上,沈阳和通辽两个环境中都检测出了显著SNPs位点(P<1.0×10-6),但未发现相同的显著SNPs位点,位于10号染色体的chr10_131199866标记(沈阳)和 chr10_130000984、chr10_130001456、chr10_130001612、chr10_130001742、chr10_130001760 5个连锁标记(通辽)最近,距离是1.20Mb。
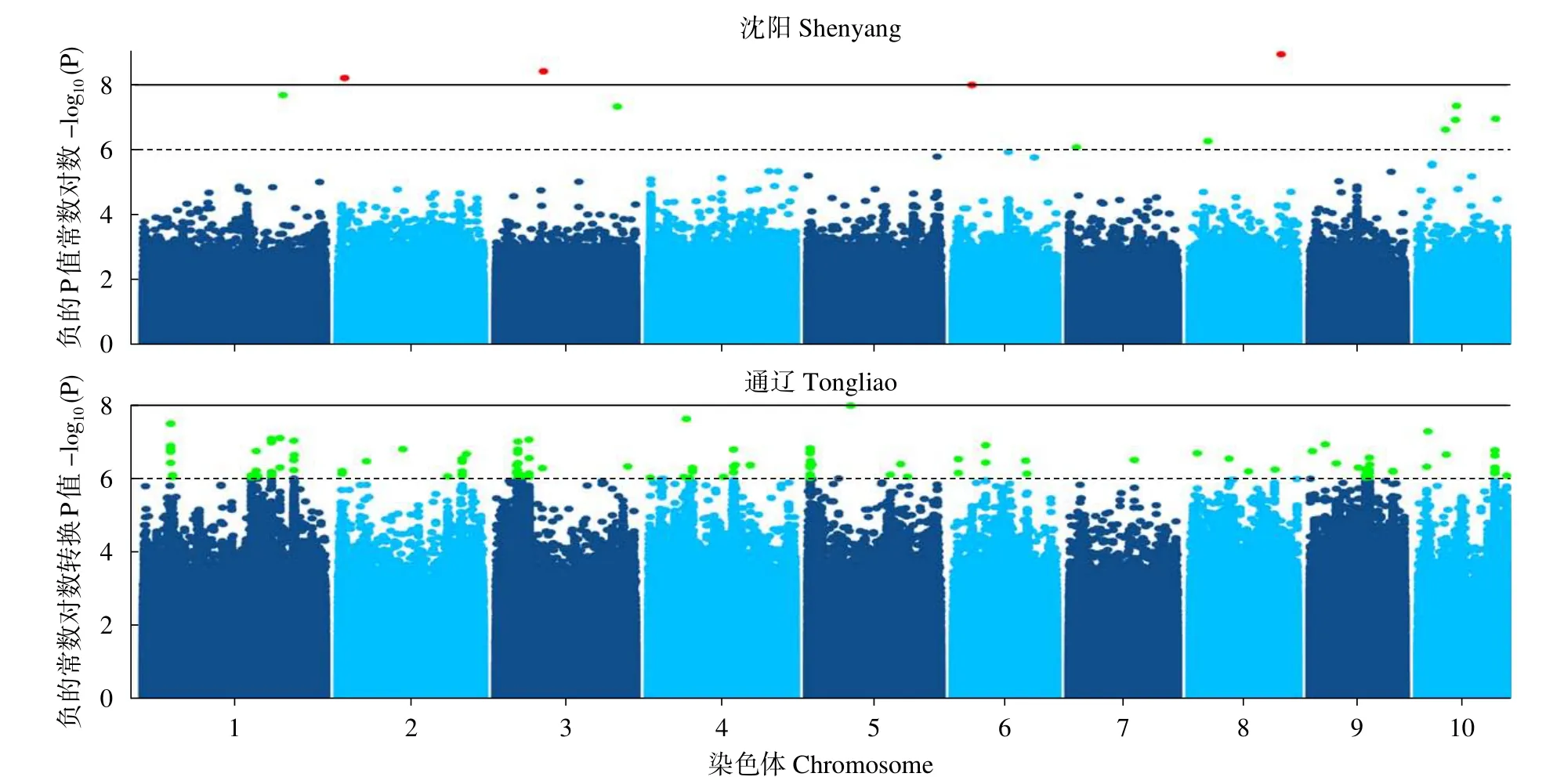
图2 不同环境(沈阳和通辽)秸秆48h IVDMD全基因组关联曼哈顿图Fig.2 Manhattan plot of genome-wide association under the environments of Shenyang and Tongliao
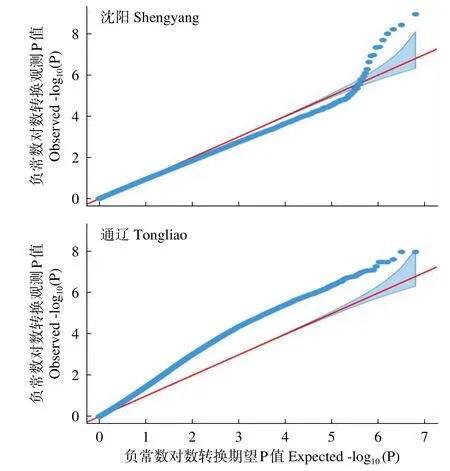
图3 不同环境秸秆48h IVDMD全基因组关联QQ图Fig.3 Quantile-quantile plot of genome-wide association under the environments of Shenyang and Tongliao
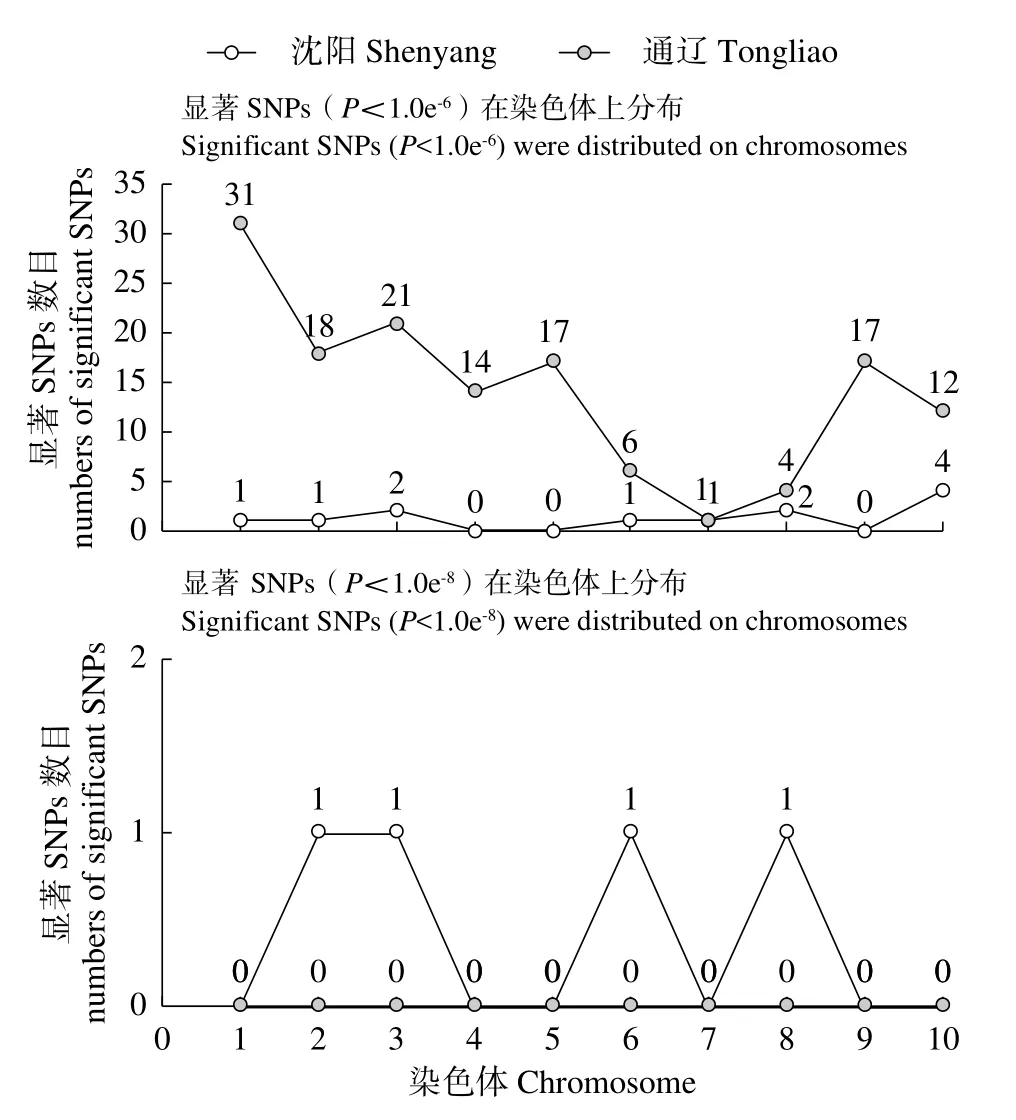
图4 显著SNPs在染色体上分布Fig.4 Significant SNPs were distributed on chromosomes
采用混合线性模型(MLM)对341份玉米自交系的48h IVDMD进行全基因组关联,共发现分布在第2、4~9染色体的10个SNPs显著位点(P<1.0×10-6),结果仍没有发现两个环境相同的SNPs显著位点(P<1.0×10-6)(图 5)。利用 MLM不仅没有关联到两个环境中和两个性状显著相关的相同的位点,而且两个环境中在共同染色体上也没有检测到显著位点。从两个模型的曼哈顿图和QQ图可以看出,FarmCPU模型关联的显著位点优于MLM的,说明FarmCPU模型关联效率要高于MLM,候选基因易从FarmCPU模型关联的显著位点中预测。
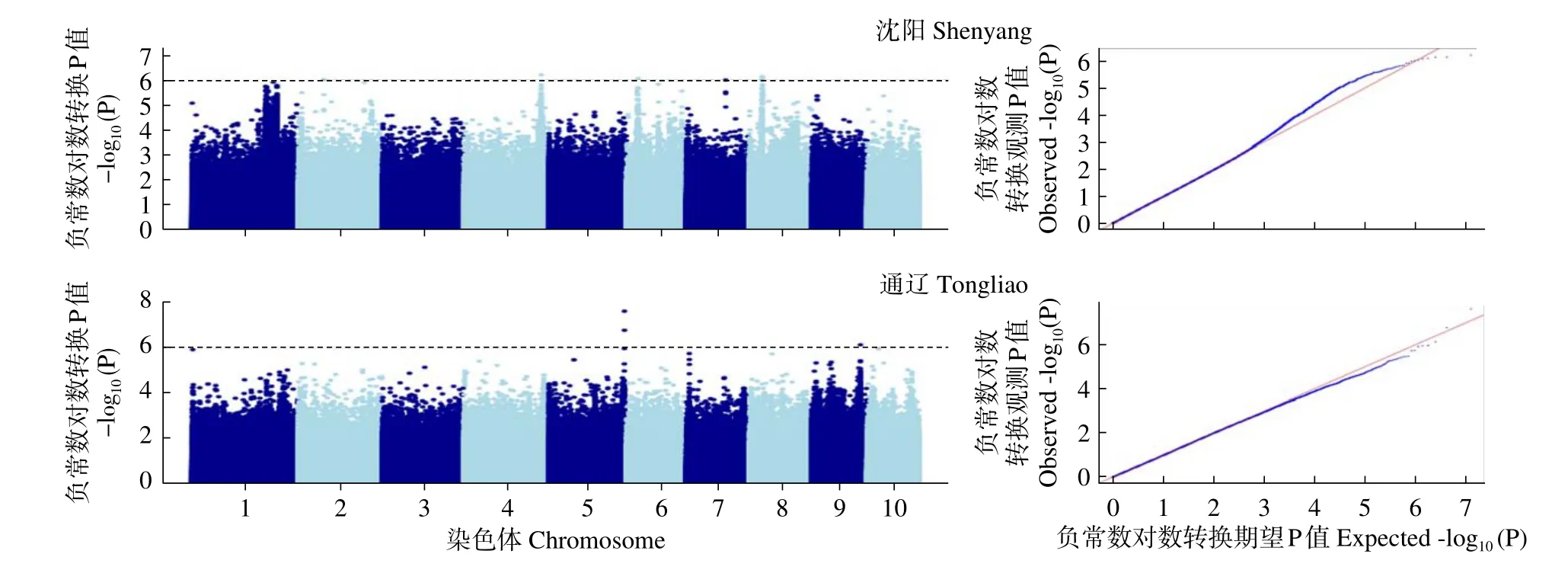
图5 两个环境秸秆48h IVDMD全基因组关联曼哈顿图及其QQ图(MLM)Fig.5 Manhattan plots and its QQ plots of genome-wide association in two environments (MLM)
2.3 候选基因分析
基于FarmCPU模型关联的153个NDF的SNPs显著位点(P<1.0×10-6)预测候选基因,结合玉米B73参考基因组组装V4注释信息,共预测到38个候选基因。在38个候选基因中,有3个候选基因编码未知功能蛋白,有1个候选基因编码MYB转录因子,其余大多数候选基因参与生长发育和防御反应的生物过程,少数候选基因涉及信号转导途径、细胞代谢途径和RNA相关途径。最重要的是,本次关联分析发现了Zm00001d009009基因和Zm00001d028874基因可能与玉米秸秆纤维素和半纤维素合成有关[19-20]。
群体变异位点分析表明,64个SNPs显著位点(P<1.0×10-6)位于38个候选基因中的34.38%内含子区、28.13%外显子区、23.44% 5'/3'非翻译区、12.50%基因上下游区、及1.56%非编码RNA区(图6)。其中,在28.13%(18个SNPs)外显子变异中,有13个非同义SNPs位于9个候选基因中,这些基因功能涉及RNA/DNA结合蛋白、开花调控、ABA信号转导负调控因子、叶和花形态建成、耐热、十八烷类防御通路[21-25]。
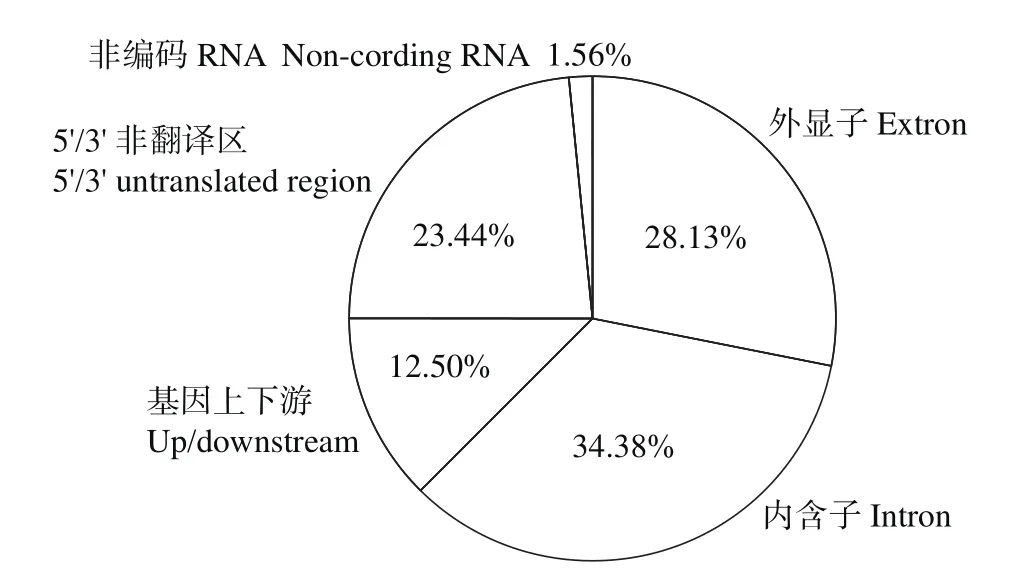
图6 变异基因的位置Fig.6 The location of the variant gene
3 讨论
3.1 GWAS定位结果分析
Li等[5]以郑58和HD568组成的220份RIL群体为材料,用6个消化性状定位了47个QTLs,其中位于2、7~10号染色体的9个QTLs控制IVDMD,解释单个QTL 5.3%的表型变异,其中定位在9.03染色体上QTL和Chr9_25266410标记最小仅距88kb。Wang等[7]以CE03005高油玉米自交系和B73组成的211份材料对玉米消化性状进行QTL研究,其中,在4~6和10号染色体上发现有6个位点与IVDMD显著相关,解释单个位点10.6%的表型变异。本试验和48h IVDMD显著相关的标记 chr4_59965077、chr4_63943611、chr4_69161939、chr4_74067130、chr4_74067534和 chr4_74243482均定位在了bin4.05染色体区段内,本试验和48h IVDMD显著相关的标记chr6_31376191、chr6_8473014、chr6_8473017、chr6_54601996和 chr6_54602105均定位在了bin6.01染色体区段内,本试验和48h IVDMD显著相关的标记chr10_46816004、ch10_63652102、chr10_65363792、chr10_15312554、chr10_17307934和chr10_48100033均定位在了bin10.03染色体区段内,与本研究结果基本一致。王琪[6]用B73和By804作亲本构建RIL群体,在3号染色体鉴定了消化率的遗传位点,本试验在这个区域也发现了相关QTL。
本试验并非所有显著SNP位点都在已知的QTL定位区间,可能原因在于:QTL作图受亲本遗传背景或检测微效QTL功效低的原因;双亲群体中的功能位点可能在关联分析群体中是稀有等位变异,因而导致在关联分析群体中不能检测;关联分析群体存在双亲群体中可能不存在的遗传变异;关于IVDMD的QTL研究非常少见。
3.2 有潜力的候选基因分析
鉴定玉米秸秆消化率的相关基因能更好地理解细胞壁合成的分子遗传机制。在本试验38个候选基因中,基因Zm00001d009009编码产物类糖基转移酶KOBITO1是高度保守的、具有植物特异性的新型质膜结合蛋白,该蛋白是拟南芥细胞扩张过程中合成纤维素所必需的蛋白。在所有光和黑暗环境中,KOB1基因突变导致拟南芥下胚轴变短,与野生型植株相比,该突变体中纤维素的相对含量下降了33%,证明该蛋白通过调节纤维素合成,促进植株下胚轴生长[26-27]。基因Zm00001d028874编码产物是木葡聚糖6-木糖基转移酶2,木葡聚糖是禾本科植物初生细胞壁半纤维素合成的主要组分,木糖基转移酶2(XXT2)将核苷酸二磷酸糖中的木糖残基转移到木聚糖主链中的葡聚糖O-6位置,研究表明,XXT2突变使木聚糖含量下降近30%,另外,木葡聚糖6-木糖基转移酶突变导致水稻根毛发育异常;有研究表明,编码木葡聚糖6-木糖基转移酶基因OsXXT1在维持水稻细胞壁结构和抗拉强度方面很重要[28-30]。
秸秆细胞壁相关性状不仅能调节玉米生长,而且在作物育种上,与抗逆性表现出很强的相关性[31],可以增强对病虫的免疫机能,提高对不利环境的适应能力。在本试验38个候选基因中,基因Zm00001d031449含有1个非同义SNP,将333位和678位的异亮氨酸突变成甲硫氨酸,编码叶绿体脂氧合酶Ⅱ。虫害的番茄叶片中TomLoxD基因上调,编码1个叶绿体LOX,该LOX可能是十八烷类防御信号通路的组成部分,该通路在一系列酶的作用下产生的茉莉酸与其受体结合,活化相应基因,产生能削弱或阻断害虫消化道内蛋白酶的蛋白酶抑制素,使害虫厌食或消化不良而死[32-33]。Zm00001d044201含有 1个非同义 SNP,将221位的丙氨酸突变成丝氨酸,编码ATP依赖Zn2+蛋白酶FTSH11,仅位于叶绿体和线粒体,研究表明,在高于30℃的环境中,拟南芥FtSH11突变体的生长发育受到抑制,而野生型植株不受影响,故该基因产物可能和植物耐热相关[34]。基因Zm00001d031449和Zm00001d044201可能因自然选择而发生非同义突变,但编码产物对植物生长和适应性存在正调节作用。基因Zm00001d043581编码产物是一类类枯草杆菌蛋白酶,与病原体识别及互作,同时被病原体诱导,启动细胞程序性死亡和超敏反应,对植物产生免疫反应[35]。综上,这些抗性基因的发现说明秸秆细胞壁是保护植株免受生物和非生物伤害的天然屏障。
秸秆消化率性状在转录水平上也受到调控。位于3'非翻译区的Zm00001d028777是ZmMs9基因,编码R2/R3植物特异性MYB转录因子,在植物次生代谢中起重要作用,与1号染色体上P1基因有关,P1基因和调节基因C1有较高的同源性,通过调节A1和C2基因,参与玉米黄酮醇生物合成过程,可能和防御紫外线相关[36-37]。
综上,玉米秸秆细胞壁是玉米健壮生长的必需结构,也是草食牲畜主要的纤维和能量来源,理解秸秆细胞壁合成机制和玉米生长适应性机制对找到有关消化率性状的基因有帮助。本文初探了青贮玉米秸秆48h IVDMD的遗传机制,为分子改良纤维含量和消化率提供了数据支撑,也为优质青贮玉米选育提供了理论参考。
4 结论
利用6 276 612个高质量SNPs(MAF>0.05)对341份玉米自交系秸秆48h IVDMD进行全基因组关联分析,两个环境中采用FarmCPU模型在所有染色体上检测到153个SNPs位点(P<1.0×10-6)和秸秆48h IVDMD显著相关,共挖掘38个候选基因,主要涉及细胞生长发育、防御反应、信号转导和细胞代谢等相关生物学功能。Zm00001d009009基因和Zm00001d028874基因可能分别与玉米秸秆纤维合成和初生壁合成有关。
猜你喜欢
湖南饲料(2019年5期)2019-10-15
科学之谜(2019年3期)2019-03-28
北京农学院学报(2019年1期)2019-02-22
科学之谜(2018年8期)2018-09-29
河南农业科学(2017年4期)2017-04-12
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
西南农业学报(2016年5期)2016-05-17
西南农业学报(2016年6期)2016-04-16
特产研究(2016年3期)2016-04-12
动物营养学报(2015年10期)2015-12-01
