
聚乙二醇修饰β-乳球蛋白羧基对其抗原性和结构的影响
2022-01-06 05:00罗舜菁熊绍百钟俊桢朱晓明江辛琳刘成梅
食品科学 2021年24期
罗舜菁,吉 莉,熊绍百,钟俊桢,朱晓明,江辛琳,刘成梅,
(1.南昌大学 食品科学与技术国家重点实验室,江西 南昌 330047;2.江西省辅食营养食品工程技术研究中心,江西 赣州 341000)
β-乳球蛋白(β-lactoglobulin,β-LG)是牛乳中主要的过敏原之一[1],降低其致敏性对牛乳过敏人群十分必要。在针对致敏性的研究中,化学方法如糖基化[2-3]、磷酸化[4]、PEG化[5]是目前国际乳制品加工研究的热点。
聚乙二醇(polyethylene glycol,PEG)修饰也称PEG化,是将活化的PEG分子通过共价键偶联到蛋白质或者多肽上,从而改变其理化性质的一种方法。由于无毒、无免疫原性、生物相容性良好[6],PEG已经用于多种蛋白质和多肽药物的修饰,以降低其免疫原性,降低酶解速度,延长半衰期,保持生物活性[7-10]。研究发现PEG链在氨基酸序列上的位置以及每个蛋白质分子上连接的PEG数目对偶联物的性质均有很大影响[11]。
本课题组前期分别针对β-LG的N-末端氨基,谷氨酰胺残基(Gln)及游离巯基进行PEG修饰,探究不同修饰位点对β-LG抗原性的影响,发现不同位点修饰后β-LG的抗原性变化大相径庭,N-末端和Gln修饰后抗原性显著降低[12-13],对游离巯基修饰后则表现为抗原性大幅度升高[14],而目前关于羧基修饰对β-LG抗原性影响的研究鲜见报道。β-LG具有多个游离羧基,是进一步探究PEG化程度对抗原性影响的理想基团。但由于羧基数目较多,控制PEG化程度难度大,且修饰产物难以分离纯化,给相关研究造成了极大困难。本研究在前期制备获得β-LG羧基单点和两点修饰产物的基础上,使用SP Sepharose Fast Flow阳离子交换柱对修饰产物分离纯化,使用十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfatepolyacrylamide gel electrophoresis,SDS-PAGE)及基质辅助激光解析电离飞行时间质谱(matrix-assisted laser desorption/ionization time of flight mass spectrometry,MALDI-TOF-MS)进行鉴定,间接竞争酶联免疫吸附测定(enzyme linked immunosorbent assay,ELISA)表征抗原性变化,游离巯基含量、内源荧光光谱、外源荧光光谱及圆二色谱表征结构变化,以探究PEG修饰和PEG化程度对β-LG抗原性和结构的影响以及二者间的关系,为调控β-LG的抗原性提供理论基础。
1 材料与方法
1.1 材料与试剂
牛β-LG、猪明胶 美国Sigma公司;单甲氧基聚乙二醇酰肼(methoxypoly (ethylene glycol) hydrazide,mPEG-Hz,5 kDa) 北京键凯科技股份有限公司;BCA蛋白试剂盒、SDS-PAGE试剂盒、辣根过氧化物酶(horseradish peroxidase,HRP)标记羊抗兔IgG二抗 北京索莱宝科技有限公司;兔抗β-LG IgG一抗、单组分超灵敏TMB显色液 北京博奥森生物技术有限公司;1-乙基-3-二甲氨基丙基碳二亚胺盐酸盐(N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride,EDC)、N-羟基硫代琥珀酰亚胺钠盐(N-hydroxysulfosucciniimide sodium,sulfo-NHS)北京阿拉丁生化科技股份有限公司;所有试剂均为分析纯,实验用水为超纯水。
1.2 仪器与设备
Mini-PROTEAN小型垂直电泳仪、NGC Quest 10 Plus中高压柱层析系统、Enrich SEC70 10 mm×300 mm凝胶柱 美国Bio-Rad公司;SP Sepharose Fast Flow色谱柱美国GE公司;Five Easy Plus pH计、ME104型电子分析天平 梅特勒-托利多仪器(上海)有限公司;Heraeus Multifuge X1R型冷冻离心机 美国Thermo公司;Amico U1tra-15超滤离心管 美国Millipore公司;MOS 405型圆二色谱仪 法国Bio-Logic公司;F-7000荧光分光光度计日本Hitachi公司;Synergy H1酶标仪 美国BioTek公司;4800 Plus MALDI-TOF/TOFTMAnalyzer质谱仪美国AB SCIEX公司。
1.3 方法
1.3.1 修饰产物的制备
羧基修饰的原理如下:
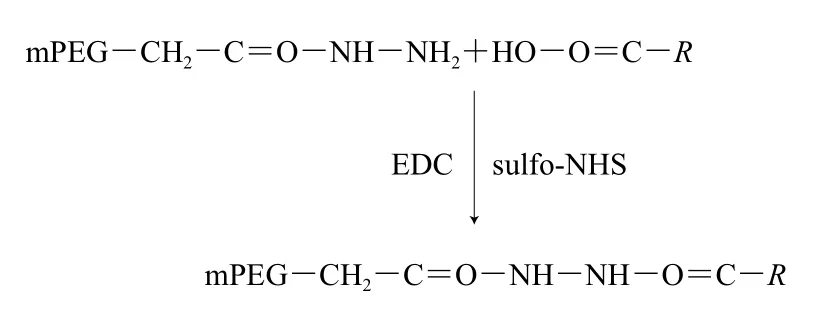
在弱酸性条件下,β-LG的氨基质子化,mPEG-Hz由于具有较低的解离常数(pKa),在EDC和sulfo-NHS的活化和助活化作用下会与蛋白质羧基选择性交联[15]。将β-LG溶于磷酸缓冲液(0.1 mol/L,pH 4.0),质量浓度为2 mg/mL,按照β-LG、EDC、sulfo-NHS物质的量比为1∶50∶10,依次加入EDC和sulfo-NHS,混合均匀后按照β-LG与PEG物质的量比为1∶20加入mPEG-Hz(5 kDa),置于4 ℃反应过夜。
1.3.2 SDS-PAGE分析
配制12%分离胶及5%浓缩胶,取30 μL反应混合物与10 μL 4×上样缓冲液(含二硫苏糖醇)混合,煮沸5~10 min后离心1~2 min,取上清液10 μL加入样品孔,以80 V电压跑浓缩胶15 min,随后以120 V的电压跑分离胶,待溴酚蓝条带至底部时停止电泳,取出胶条于考马斯亮蓝染色液中染色1 h后脱色至蛋白条带清晰。
1.3.3 阳离子交换色谱分析
参考Zhong Junzhen等[5]的方法,使用SP Sepharose Fast Flow阳离子交换柱对修饰产物分离纯化。使用3 倍柱体积的醋酸钠缓冲液(0.04 mol/L,pH 4.0)对离子柱平衡后上样,使用含1.0 mol/L NaCl的醋酸钠缓冲液(0.04 mol/L,pH 4.0)0%~100%对样品线性梯度洗脱150 min,流速2 mL/min,在280 nm波长处测定吸光度。产物的修饰率通过层析系统自带软件计算,计算如式(1)所示:

1.3.4 凝胶过滤色谱分析
使用Enrich SEC70 10 mm×300 mm凝胶柱对修饰产物纯度进行分析。使用磷酸盐缓冲液(10 mmol/L,pH 7.0)平衡凝胶柱,待基线水平且电导率保持不变后,上样0.2 mL进行洗脱,流速1 mL/min,于280 nm波长处检测吸光度。
1.3.5 MALDI-TOF-MS分析
使用4800 Plus MALDI-TOF/TOF质谱仪对修饰前后β-LG的分子质量进行分析鉴定。取1 μL样品点至样品靶上自然干燥,再取1 μLα-氰基-4-羟基肉桂酸基质溶液点至对应靶位上自然干燥,同样的方法在相邻靶位上点校准标准品(4700 Proteomics Analyzer Calibration Mixture 1)。采用校准标准品进行外标一级校准,波长349 nm,电压2 kV,在正离子,线性模式下进行检测,扫描范围5 000~50 000 Da,质量误差小于0.5 Da,数据通过软件Data Explorer V4.5进行分析。
1.3.6 抗原性分析
参考Meng Xuanyi等[16]的方法,用间接竞争ELISA法对修饰产物与抗体的结合能力进行分析测定,通过半抑制浓度(IC50)表征产物抗原性变化。1)包被:在96 孔酶标板中每孔加入100 μLβ-LG(2.5 μg/mL)作为包被抗原,4 ℃过夜;2)封阻:次日用含0.05% Tween 20的磷酸盐缓冲溶液(phosphate-buffered saline Tween-20,PBST)重复洗涤扣干3 次,每孔加入250 μL 1%猪明胶进行封阻,37 ℃孵育1 h;3)竞争抑制:另一酶标板以同一方法封阻,洗涤后每孔分别加入60 μL不同质量浓度(0.1、2.5、5、10、15、25、50 μg/mL)的竞争抗原(mono-PEG-β-LG、di-PEG-β-LG、未修饰β-LG)和等体积兔抗β-LG IgG一抗(1∶35 000稀释),用PBST代替竞争抗原作空白对照,37 ℃孵育1 h,每孔取100 μL转移至第一块板内进行竞争,37 ℃孵育1 h;4)二抗反应:洗涤扣干后每孔加入HRP标记的羊抗兔IgG酶标二抗(1∶5 000稀释)100 μL,37 ℃孵育1 h;5)显色终止:洗涤后每孔加100 μL单组分TMB显色液,37 ℃避光25 min后加入等体积终止液终止反应;6)检测计算:5 min后于450 nm波长处检测OD值。以修饰产物浓度为横坐标,抑制率为纵坐标,计算IC50值,表征修饰产物抗原性变化。抑制率计算如式(2)所示:
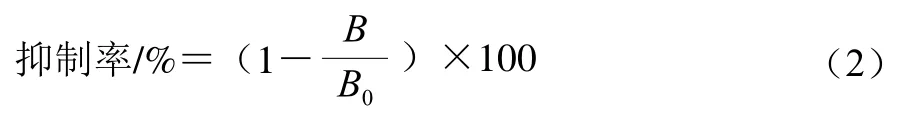
式中:B为竞争抗原对应的OD值;B0为空白对照孔的OD值。
1.3.7 游离巯基含量分析
使用Ellmans’ DTNB法[17]对修饰前后样品的游离巯基含量进行测定。取1 mL待测蛋白溶液(1.0 mg/mL)加入到4 mL Tris-Gly缓冲溶液(0.086 mol/L Tris、0.09 mol/L Gly、5 mmol/L EDTA,pH 8.0)中,加入50 μL Ellmans’试剂(4 mg/mL),37 ℃反应20 min后使用Synergy H1酶标仪于412 nm波长处检测吸光度。游离巯基含量计算如式(3)所示:
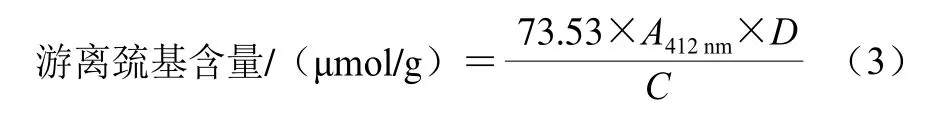
式中:A412nm为样品在412 nm处的吸光度;D为稀释倍数;C为样品质量浓度/(mg/mL)。
1.3.8 表面疏水性分析
以1-氨基萘-8-磺酸(1-anilinonaphthalene-8-sulfonate,ANS)为荧光探针,使用荧光分光光度计对修饰前后β-LG的表面疏水性进行分析。PBS溶液(10 mmol/L,pH 7.0)稀释样品至0.1 mg/mL,取4 mL样品与20 μL ANS(8 mmol/L)混合,避光反应5~10 min,激发波长370 nm,发射波长400~600 nm,狭缝宽度5 nm,扫描速率1 200 nm/min,以PBS替代样品作空白对照。
1.3.9 内源荧光光谱分析
使用荧光分光光度计分析修饰前后β-LG内源荧光强度的变化。10 mmol/L PBS溶液(pH 7.0)稀释样品至0.1 mg/mL,激发波长为280 nm,扫描发射波长范围300~450 nm,狭缝宽度2.5 nm,扫描速率1 200 nm/min,重复扫描3 次。
1.3.10 圆二色谱分析
使用圆二色谱仪对修饰产物的二级结构组成及含量进行分析。样品质量浓度0.1 mg/mL,使用石英比色皿于25 ℃在氮气环境下检测,扫描范围190~250 nm,步进分辨率1 nm,谱带宽度1 nm,扫描速率100 nm/min,重复扫描3 次。通过在线网站Dichroism Website分析拟合圆二图谱并计算二级结构相对含量。
1.4 数据统计与分析
使用Origin 8.5软件作图,SPSS 17.0软件分析数据显著性(P<0.05,差异显著)。
2 结果与分析
2.1 产物分离纯化及修饰率检测
等电点是离子交换层析的重要依据。β-LG分子质量为18.3 kDa,等电点在5.1~5.3之间[18],在醋酸钠(40 mmol/L,pH 4.0)缓冲体系中带正电,与带负电的阳离子交换树脂结合。使用含1.0 mol/L NaCl的醋酸钠缓冲液(40 mmol/L,pH 4.0)在0%~100%进行线性梯度洗脱,结果如图1A所示,得到3 个明显的洗脱峰F1、F2和F3。带正电的β-LG与不带电的PEG分子结合减弱了蛋白质与吸附表面之间的静电作用,使其与离子柱结合能力降低,PEG化程度越高,结合能力越弱[19],洗脱所需的盐浓度越低,因而PEG修饰产物先于未修饰的β-LG被洗脱。推测峰F1和F2为两种修饰产物的洗脱峰,且F1的PEG化程度高于F2,F3为未修饰的β-LG。
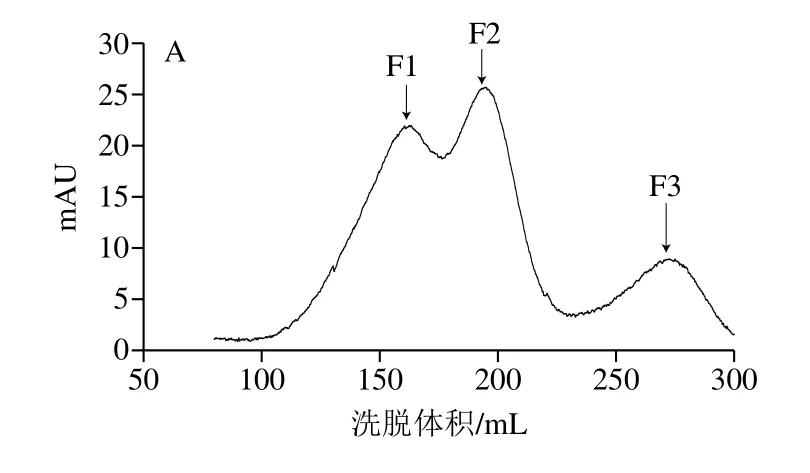
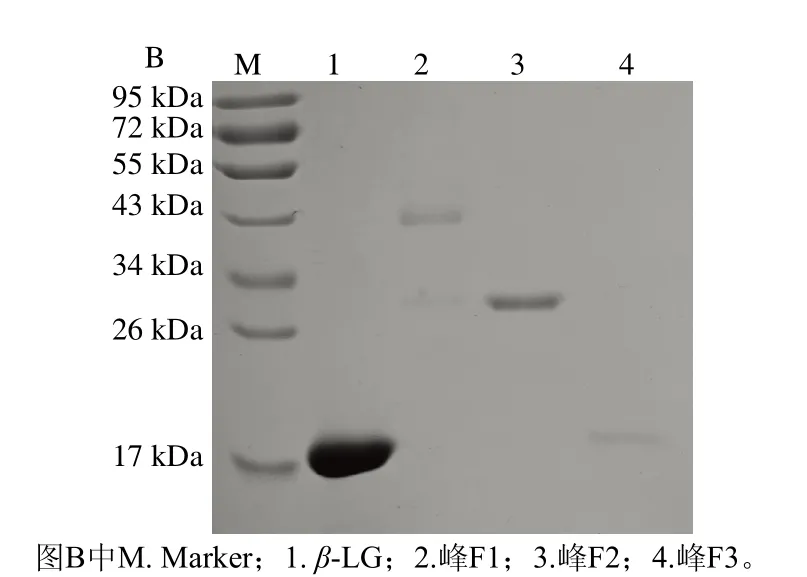
图1 PEG修饰混合物的阳离子交换色谱图(A)和纯化得到的各洗脱峰SDS-PAGE图(B)Fig.1 Cation exchange chromatogram of PEGylation mixture (A) and SDS-PAGE analysis of elution peaks obtained from purification (B)
本研究采用的mPEG-Hz会在EDC和sulfo-NHS的助活化作用下与蛋白质羧基选择性交联。收集洗脱峰进行SDS-PAGE分析,如图1B所示,峰F3位于18 kDa左右,与β-LG分子质量相同,保留时间最短的峰F1在34~43 kDa之间有一条带,F2在26~34 kDa之间有一条带,修饰后β-LG分子质量显著增加,通过层析系统自带软件分析,产物的总修饰率为82.91%。
2.2 修饰产物纯度分析
因F1和F2未完全分开,分别大量收集F1峰前及F2峰后,用截留分子质量10 kDa的Amico Ultra-15超滤离心管于4 ℃、6 000 r/min离心20 min,用10 mmol/L PBS(pH 7.0)对样品中溶液进行置换。利用凝胶柱的分子筛作用,各物质按照分子质量递减顺序洗脱,对修饰产物纯度进行鉴定。如图2所示,两种产物的洗脱峰均窄而尖,峰F1的浓缩样品纯度高达98.61%(图2A),峰F2的浓缩样品纯度高达99.66%(图2B),表明成功制备纯度均在95%以上的两种PEG修饰产物。
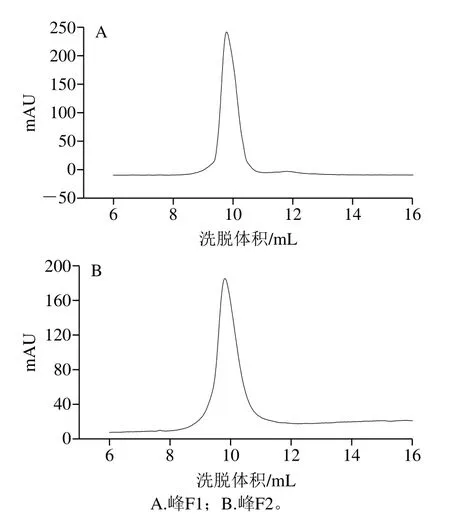
图2 PEG修饰产物的凝胶过滤色谱图Fig.2 Gel filtration chromatogram of PEGylation products
2.3 修饰产物的鉴定
2.3.1 SDS-PAGE分析
大量收集两种修饰产物并浓缩,稀释4 倍后进行SDS-PAGE分析。如图3所示,泳道2和3分别在26~34 kDa之间和34~43 kDa之间具有一条明显的条带,表明阳离子交换柱分离效果较好。
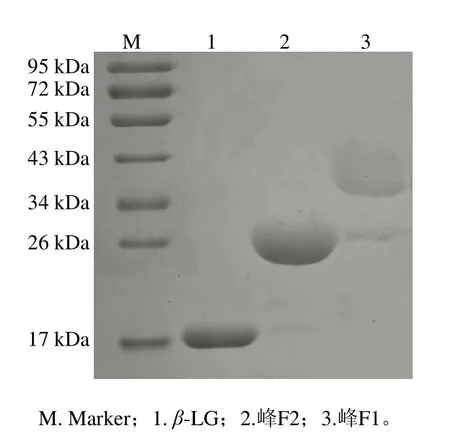
图3 PEG修饰产物的SDS-PAGE图Fig.3 SDS-PAGE analysis of PEGylation products
溶液中PEG分子的乙氧基会结合水分子,使PEG修饰产物的流体力学体积增大,因而修饰产物的表观分子质量是未修饰蛋白的分子质量与2.5 倍PEG分子质量之和[20],泳道2的表观分子质量是β-LG分子质量与单个2.5 倍PEG分子质量(5 kDa)之和,泳道3对应的是β-LG分子质量与两个2.5 倍PEG分子质量(5 kDa)之和,表明泳道2(峰F2)和3(峰F1)分别为mono-PEG-β-LG和di-PEG-β-LG。
2.3.2 MALDI-TOF-MS分析
为进一步鉴定两种修饰产物,使用MALDI-TOF-MS对β-LG及修饰产物的实际分子质量进行测定。
如图4所示,β-LG标准品分子质量为18.3 kDa(图4A),两种修饰产物的分子质量分别为23.3 kDa(β-LG分子质量与一个PEG分子质量之和)和28.6 kDa(β-LG分子质量与两个PEG分子质量之和),进一步证明纯化得到的PEG修饰产物为mono-PEG-β-LG和di-PEG-β-LG。
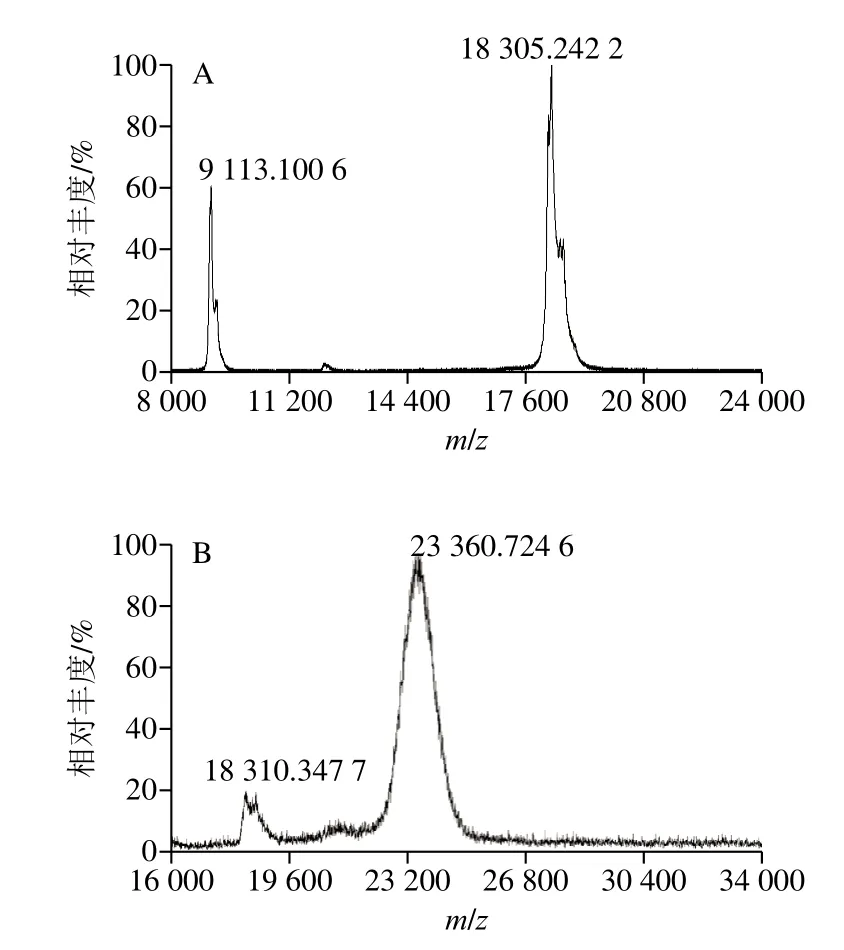
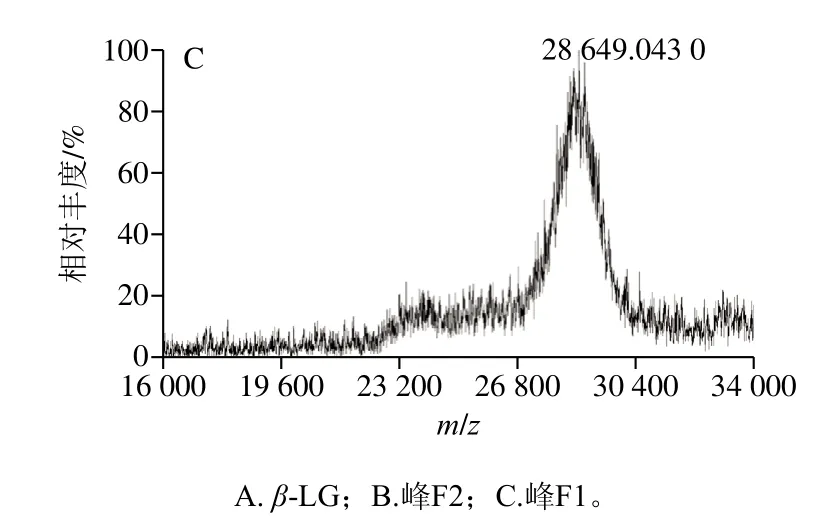
图4 PEG修饰产物的MALDI-TOF-MS图Fig.4 MALDI-TOF-MS analysis of PEGylation products
2.4 抗原性检测结果
在间接竞争ELISA中,使用IC50值表征修饰产物与抗体的结合能力,IC50值越高,表明达到IC50值所需竞争抗原越多,即竞争抗原与抗体的结合能力越弱,修饰产物抗原性越弱[21]。如图5所示,β-LG的IC50值最低,为1.90 μg/mL,mono-PEG-β-LG的IC50值为2.47 μg/mL,是β-LG的1.30 倍,而di-PEG-β-LG高达10.41 μg/mL,为β-LG的5.48 倍,表明PEG修饰后β-LG的抗原性显著降低,且提高PEG化程度可以极显著降低其抗原性。
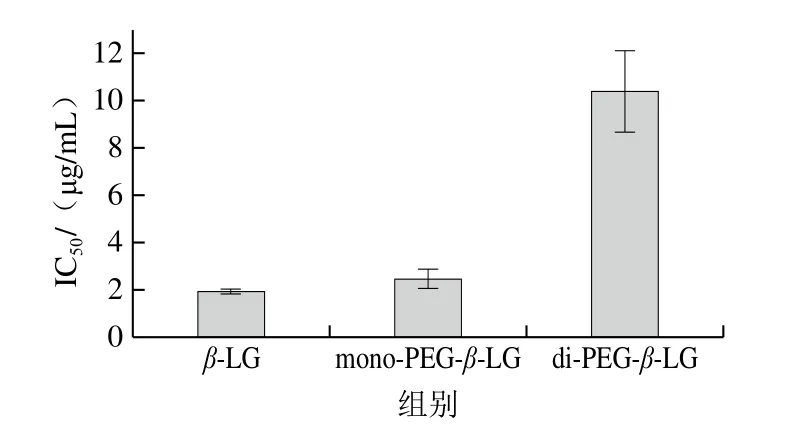
图5 PEG修饰产物的抗原性变化图Fig.5 Changes in antigenicity of PEGylation products
研究表明,β-LG的抗原性取决于线性致敏表位和构象致敏表位,而致敏表位与结构变化密切相关[22]。本团队前期研究发现,游离巯基修饰后β-LG抗原性升高的可能原因是由于结构变化导致新致敏表位形成[14],而N-末端及Gln修饰后抗原性降低的可能原因是PEG分子遮蔽了β-LG抗原性表位[12,23]。为明确羧基修饰后β-LG抗原性降低的原因,实验需对修饰产物的结构变化进行分析。
2.5 游离巯基含量分析
巯基对维持蛋白质结构意义重大,每个β-LG单体含有5 个半胱氨酸残基(Cys),其中Cys66和Cys160、Cys106和Cys160分别形成两个二硫键,而游离巯基Cys121包埋于分子内部[24]。修饰产物游离巯基含量如图6所示,PEG修饰后,β-LG的游离巯基含量由52.54 μmol/g分别下降至42.70 μmol/g(mono-PEG-β-LG)和37.97 μmol/g(di-PEG-β-LG)。由于PEG链具有空间屏蔽作用[25],可能会对Cys121造成遮蔽,PEG化程度越高,遮蔽作用越强,检测到的游离巯基含量越低,抗原性降低越显著。
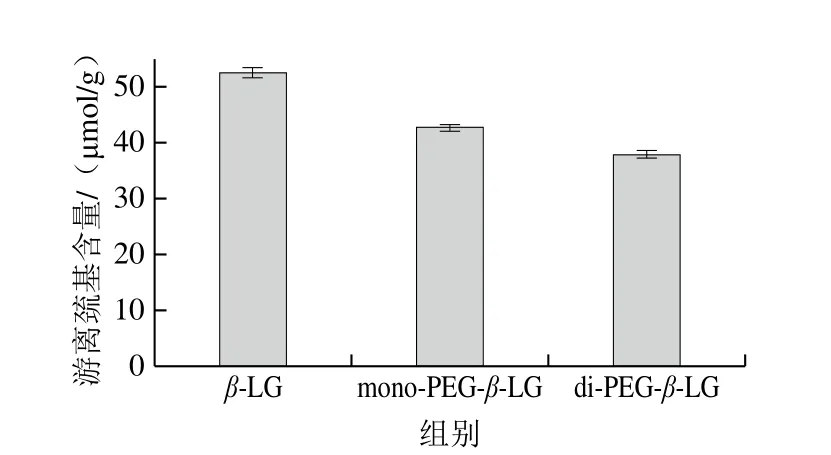
图6 PEG修饰产物的游离巯基含量分析Fig.6 Free sulfhydryl content analysis of PEGylation products
2.6 表面疏水性分析
表面疏水性与蛋白质的构象、稳定性及抗原性变化密切相关[26]。以ANS为荧光探针,通过与蛋白质表面暴露的疏水性氨基酸结合表征表面疏水性变化[16]。如图7所示,β-LG的最大荧光强度为1 821,两种修饰产物荧光强度分别为2 067和2 114,表明PEG修饰后β-LG的表面疏水性增强,且di-PEG-β-LG略高于mono-PEG-β-LG。
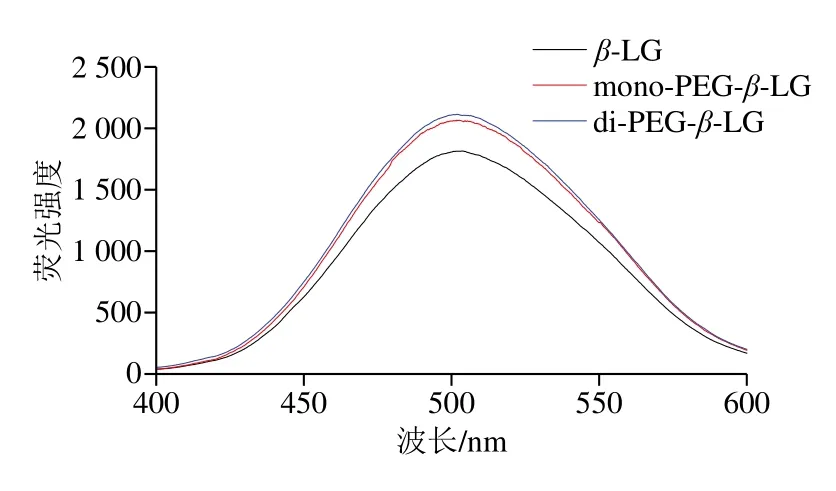
图7 PEG修饰产物的表面疏水性分析Fig.7 Surface hydrophobicity analysis of PEGylation products
β-LG分子内部具有大量疏水性氨基酸[27],在PEG结合过程中,β-LG三级结构展开,包埋于分子内部的疏水性氨基酸暴露于极性溶液中,使ANS与蛋白质的结合能力提高[28],导致表面疏水性增强;另一方面,PEG链具有很强的遮蔽作用[19],因此表现为mono-PEG-β-LG与di-PEG-β-LG表面疏水性没有显著区别。
2.7 内源荧光光谱分析
在内源荧光光谱分析中,常以色氨酸(Trp)为荧光探针,通过描述其所处微环境考察蛋白质结构变化。修饰后β-LG内源荧光光谱变化如图8所示,β-LG在333 nm波长处最大内源荧光强度为2 495,mono-PEG-β-LG和di-PEG-β-LG的内源荧光强度分别为2 382和2 340,随PEG化程度的升高,修饰产物荧光强度降低,最大发射波长未发生明显红移或蓝移。
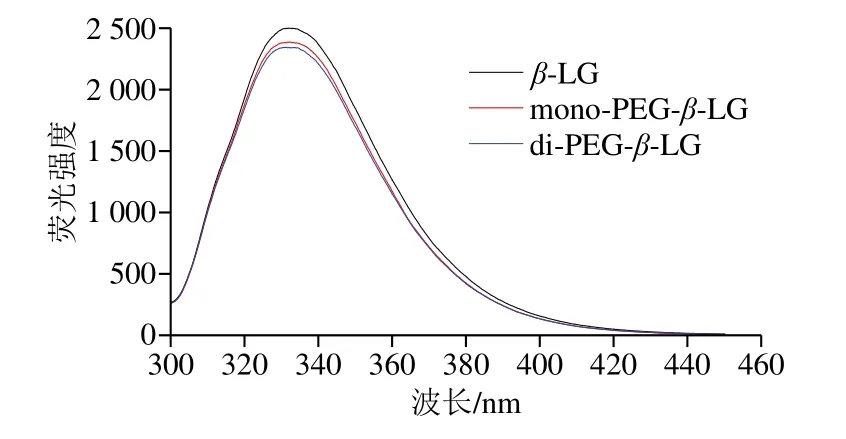
图8 PEG修饰产物的内源荧光光谱分析Fig.8 Intrinsic fluorescence spectra of PEGylation products
研究表明,位于疏水环境中的Trp暴露于极性溶剂中会发生荧光猝灭[29],因此PEG的接入可能导致β-LG的三级结构展开,分子内部19Trp和61Trp暴露于溶剂中[30],使荧光强度降低。mono-PEG-β-LG与di-PEG-β-LG的荧光强度没有显著区别,可能是由于尽管后者Trp暴露更多,但多个PEG链的屏蔽作用导致荧光强度变化不大。三级结构展开造成构象表位破坏,以及PEG的空间位阻效应导致修饰产物抗原性降低。
2.8 二级结构分析
通过圆二色谱分析PEG修饰对β-LG二级结构的影响,结果如图9所示。β-LG是一个以β-折叠为主的蛋白,8 条反向平行的β-折叠在蛋白疏水内部形成桶状结构,桶外是一条具有3 个β-转角的α-螺旋和第9条β-折叠链[31]。192 nm波长处的正吸收峰、208 nm和222 nm波长附近的负吸收峰是α-螺旋的显著特征;190~200 nm波长处的正谱带及216~218 nm波长处的负峰是β-折叠的显著特征。
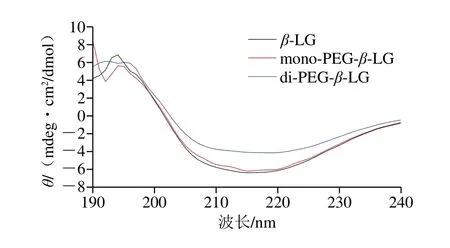
图9 PEG修饰产物的二级结构变化Fig.9 Circular dichroism spectra of PEGylation products
如表1所示,与未修饰的β-LG相比,mono-PEG-β-LG的二级结构没有发生显著变化;而di-PEG-β-LG的二级结构发生变化,表现为α-螺旋相对含量显著降低,无规卷曲相对含量显著升高,说明在di-PEG-β-LG形成过程中β-LG的二级结构展开,构象致敏表位破坏程度加剧。

表1 PEG修饰产物的二级结构相对含量变化Table 1 Changes in secondary structure contents in PEGylated products%
3 结 论
采用mPEG-Hz(5 kDa)对β-LG羧基共价修饰,制备不同PEG化程度的产物,总修饰率为82.91%。对修饰产物进行分离纯化,获得纯度分别为99.66%和98.61%两种主要修饰产物;SDS-PAGE及MALDI-TOF-MS鉴定产物分子质量分别为23.3 kDa和28.6 kDa,表明修饰产物分别为mono-PEG-β-LG及di-PEG-β-LG。
β-LG、mono-PEG-β-LG和di-PEG-β-LG的IC50分别为1.90、2.47 μg/mL和10.41 μg/mL,mono-PEG-β-LG和di-PEG-β-LG的IC50值分别是β-LG的1.30 倍和5.48 倍,表明羧基修饰后β-LG的抗原性显著降低,且提高PEG化程度可以极显著降低β-LG的抗原性。两种修饰产物的游离巯基含量分别为42.70 μmol/g和37.97 μmol/g,表明随着PEG化程度增强,结合的PEG分子增多,遮蔽作用增强;相比于mono-PEG-β-LG仅发生三级结构的变化,di-PEG-β-LG的二级结构也发生变化(α-螺旋相对含量显著降低,无规卷曲相对含量显著升高),表明随着PEG化程度增强,β-LG的空间结构改变,可能导致构象表位破坏加剧。在PEG分子的遮蔽和空间位阻效应以及构象致敏表位破坏两重作用下,di-PEG-β-LG的抗原性发生了极显著的降低。
因此后续研究可以通过以下两种方法进一步降低β-LG的抗原性:1)通过条件优化等方法提高PEG化程度,增强对线性致敏表位的遮蔽及空间位阻作用;2)使用物理化学等方法改变β-LG结构,加剧破坏构象致敏表位。为深入探究β-LG抗原性变化的机理,还需对PEG结合位点进行分析鉴定,明晰抗原性变化与结构变化,结合位点之间的关系。
猜你喜欢
广州化学(2022年4期)2022-09-01
实用手外科杂志(2022年2期)2022-08-31
阅读(科学探秘)(2021年8期)2021-09-01
中国金属通报(2021年20期)2021-03-11
科学导报·学术(2020年29期)2020-10-21
照明工程学报(2020年1期)2020-06-16
美与时代·美术学刊(2019年9期)2019-11-29
商品与质量(2019年31期)2019-11-28
分析科学学报(2016年2期)2016-10-15
中国医疗美容(2015年4期)2015-04-27
