
六味地黄苷糖片多糖部位相对分子质量与组成分析
2022-01-27 14:41郝晓娟胡军华王振中伟1
中成药 2022年1期
郝晓娟, 付 娟, 胡军华, 王振中, 肖 伟1,*
(1.南京中医药大学,江苏 南京 210000;2.中药制药过程新技术国家重点实验室,江苏 连云港 222001;3.江苏康缘药业股份有限公司,江苏 连云港 222001)
六味地黄方由熟地黄、酒萸肉、牡丹皮、山药、茯苓、泽泻组成,始见于《小儿药证直诀》,由宋朝名医钱乙首创,是中医滋补肾阴的经典名方[1]。六味地黄苷糖片是在六味地黄汤的基础上,结合现代药理学、药效学活性评价,经两次醇沉、两步层析提取分离,由水溶性多糖、以甘露三糖为主的寡糖,以莫诺苷、马钱苷、芍药苷为主的糖苷类组成的创新中药,其中多糖部位主要表现为免疫促进作用[2]。
多糖是一类由多羟基醛或多羟基酮及其衍生物、聚合物的总称,为中药中主要药效物质基础之一[3],其分子量大,结构复杂,生物活性有明显差异,近年来越来越受到关注。由于该成分生物活性、分子量与其分布和组成密切相关,故2015年版《中国药典》已将相对分子质量及其分布列入该成分质量标准中[4-5]。
本实验以六味地黄苷糖片中多糖部位为研究对象,对其相对分子质量及组成进行初步考察,不仅为进一步研究该部位生物活性与其一级结构的关系提供基础,又为提升该制剂质量标准提供依据。
1 材料
AL204、MS-105DU电子分析天平(瑞士Mettler-Toledo公司);HH-8数显恒温水浴锅(金坛市康华电子仪器制造厂);H1850R台式高速冷冻离心机(湖南湘仪实验室仪器开发有限公司);LC-28AB高效液相色谱仪,配置RID-10A示差折光检测器(日本岛津公司);KQ-500DB型数控超声波清洗器(昆山市超声仪器有限公司);DZF-6050型真空干燥箱(上海新苗医疗器械制造有限公司);HGC-36A型氮吹仪(天津市恒奥科技发展有限公司);DHG-9070A型电热恒温鼓风干燥箱(上海精宏实验设备有限公司);Agilent7890B-7698A气相色谱仪(美国安捷伦公司);磷酸二氢钾为色谱纯,购自上海麦克林生化科技有限公司;三氟乙酸为色谱纯,购自美国天地公司;乙醇、丙酮、乙醚、吡啶、甲醇、硼氢化钠、三氯甲烷等均为分析纯,购自国药集团化学试剂有限公司;正丙胺购自阿拉丁试剂(上海)有限公司。各相对分子质量葡聚糖(T670、T410、T270、T110、T70、T40、T20、T5)购自美国Sigma-Aldrich公司、阿拉丁试剂(上海)有限公司;L-(+)-阿拉伯糖(批号WXBB0 276 V,纯度≥99%)、L-鼠李糖(批号WXBB5 979 V,纯度≥99%)均购自美国Sigma-Aldrich公司;D-无水葡萄糖(批号110833-201908,纯度99.9%)、D-木糖(批号111508-201605,纯度99.9%)、D-甘露糖(批号140651-201805,纯度99.9%)、肌醇(批号190077-201501,纯度99.6%)、半乳糖(批号100226-201807,纯度100%)、D-葡萄糖醛酸(批号140648-201804,纯度99.8%)、D-半乳糖醛酸(批号111646-201702,纯度95.1%)均购自中国食品药品检定研究院。
六味地黄苷糖片多糖部位(批号Z160601、Z171001、Z191001、Z191201、Z191202),购自江苏康缘药业股份有限公司。
2 方法
2.1 分子量测定
2.1.1 色谱条件与系统适用性试验 TSK-GEL G4000PWXL、TSK-GEL G1000PW色谱柱(7.8 mm×300 mm)串联[6-8];流动相0.02 mol/L KH2PO4溶液;体积流量0.5 mL/min;RID检测器;检测器、柱温箱温度40 ℃;进样量20 μL。
2.1.2 多糖部位前处理 参考文献[9]报道。取多糖部位适量,60%乙醇洗涤,每次20 mL,涡流2 min后离心,重复以上操作至醇洗液呈无色(约洗涤16次),转移至布氏漏斗中,分别用乙醇、丙酮、乙醚反复洗涤,挥干,备用。
2.1.3 供试品溶液制备 精密称取“2.1.2”项下醇洗多糖2.5 g,置于烧瓶中,精密加入流动相100 mL,称定质量,100 ℃下回流提取1 h,放冷,加水补足减失的质量,摇匀,14 000 r/min离心5 min,取上清液,即得。
2.1.4 对照品溶液制备 精密称取不同相对分子质量的葡聚糖、D-无水葡萄糖对照品各50 mg,置于10 mL量瓶中,加水溶解后定容至刻度,摇匀,离心,即得。
2.1.5 方法学考察
2.1.5.1 线性关系考察 取“2.1.4”项下对照品溶液,在“2.1.1”项色谱条件下进样测定。采用GPC软件,以对照品重均分子量的对数(logMw)为纵坐标(Y),保留时间为横坐标(X)进行回归,得到方程为Y=2.622 4×10-3X3-0.220 8X2+5.811 8X-43.186 6 (R2=0.995 2),在5 000~670 000 Da范围内线性关系良好。
2.1.5.2 精密度试验 精密吸取“2.1.3”项下供试品溶液20 μL,在“2.1.1”项色谱条件下进样测定6次,采用GPC软件测得峰面积RSD为0.06%,表明仪器精密度良好。
2.1.5.3 稳定性试验 精密吸取“2.1.3”项下供试品溶液20 μL,于0、3、6、9、12、15、18、21、24 h在“2.1.1”项色谱条件下进样测定,采用GPC软件测得峰面积RSD为0.49%,表明溶液在24 h内稳定性良好。
2.1.5.4 重复性试验 精密称取多糖部位样品6份,按“2.1.2”项下方法处理,按“2.1.3”项下方法制备供试品溶液,在“2.1.1”项色谱条件下进样20 μL测定,采用GPC软件测得峰面积RSD为0.81%,表明该方法重复性良好。
2.1.6 重均分子量测定 取六味地黄苷糖片多糖部位Z160601、Z171001、Z191001、Z191201、Z191202进行测定,测得重均相对分子质量分别为113 912、146 446、151 939、191 694、177 122。
2.2 多糖组成分析
2.2.1 气相色谱条件 ZB-5色谱柱(30 m×0.25 mm×0.25 μm);氢火焰检测器,进样口温度250 ℃,检测器温度280 ℃;氮气体积流量0.6 mL/min;分流比20∶1;程序升温(200 ℃保持2 min,3 ℃/min升至245 ℃,10 ℃/min升至270 ℃,保持2 min)[10-13];进样量2 μL。
2.2.2 标准品衍生物制备 精密称取鼠李糖、阿拉伯糖、木糖、肌醇、甘露糖、无水葡萄糖、半乳糖、葡萄糖醛酸、半乳糖醛酸对照品各2 mg,置于10 mL具塞试管中,加入1 mL水溶解,再加入0.5 mol/L 碳酸钠溶液78 μL,30 ℃下水浴45 min,加入4%硼氢化钠溶液0.5 mL,室温下放置1.5 h,滴加25%乙酸至无气泡产生以除掉过量硼氢化钠,将反应液过阳离子交换树脂柱,并用8 mL水洗脱,洗脱液浓缩至干后加入3 mL甲醇,浓缩至干,重复5次以除净过量乙酸。将浓缩至干的样品于85 ℃下真空干燥2 h,加吡啶、正丙胺各1 mL,55 ℃下水浴30 min,放冷后于55 ℃水浴中氮气吹干,加吡啶、乙酸酐各0.5 mL,95 ℃下水浴1 h,放冷后于25 ℃水浴中氮气吹干,于40 ℃下真空干燥3 h,取无水硫酸钠干燥24 h 的0.3 mL氯仿溶解样品,取10 μL,氯仿稀释至200 μL,即得,并进行GC分析[14-15]。
2.2.3 样品衍生物制备 精密称取样品5 mg,加入0.4 mol/L三氟乙酸3 mL,100 ℃下水浴4 h,加入甲醇3 mL浓缩至干,反复4~5次以除净三氟乙酸,加1 mL水充分溶解,从“加入0.5 mol/L 碳酸钠溶液78 μL”开始,其余操作同“2.2.2”项下,即得。
2.2.4 单糖组成测定 取六味地黄苷糖片多糖部位Z160601、Z171001、Z191001、Z191201、Z191202进行测定,结果见表1。
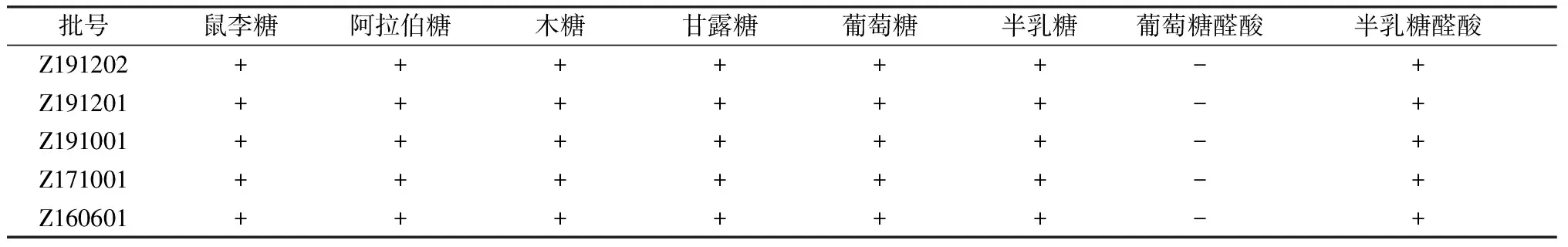
表1 单糖组成测定结果
3 结果
3.1 相对分子质量测定
3.1.1 流动相选择 取“2.1.3”项下供试品溶液、“2.1.4”项下对照品溶液,分析采用TSK-GEL G4000PWXL(7.8 mm×300 mm)色谱柱;流动相Na2SO4、NaH2PO4、KH2PO4溶液;体积流量0.5 mL/min;RID检测器、柱温箱温度40 ℃,结果见图1。由此可知,Na2SO4溶液洗脱时供试品溶液在保留时间20.5 min附近有1个固定倒峰,可影响葡聚糖出峰,而NaH2PO4、KH2PO4溶液洗脱时不存在倒峰,故选择磷酸盐溶液作为流动相。

图1 各流动相下T40 HPGPC色谱图
3.1.2 供试品提取方式选择 取“2.1.2”项下多糖样品,加流动相涡流2 min,制成每1 mL含25 mg单糖的供试品溶液,离心,取上清液,在“2.1.1”项色谱条件下进样测定,结果见图2。由此可知,多糖样品直接涡流后进样时色谱图分离度较差,重均相对分子质量20 000~70 000,可能是因为涡流不能将大分子量的多糖提取完全。

图2 涡流提取多糖分子量HPGPC色谱图
3.1.3 串联色谱柱选择 固定流动相为KH2PO4溶液,检测器、柱温箱温度均为40 ℃,体积流量0.5 mL/min,RID检测器,考察单用TSK-GEL G4000PWXL色谱柱(7.8 mm×300 mm)及串联TSK-GEL G1000PW色谱柱(7.8 mm×300 mm)的分离效果,结果见表2、图3~4。由此可知,单根色谱柱的分离度低于串联色谱柱,不能很好地表明多糖部位的重均相对分子质量,故本实验选择串联色谱柱。

表2 单根色谱柱分子量测定结果
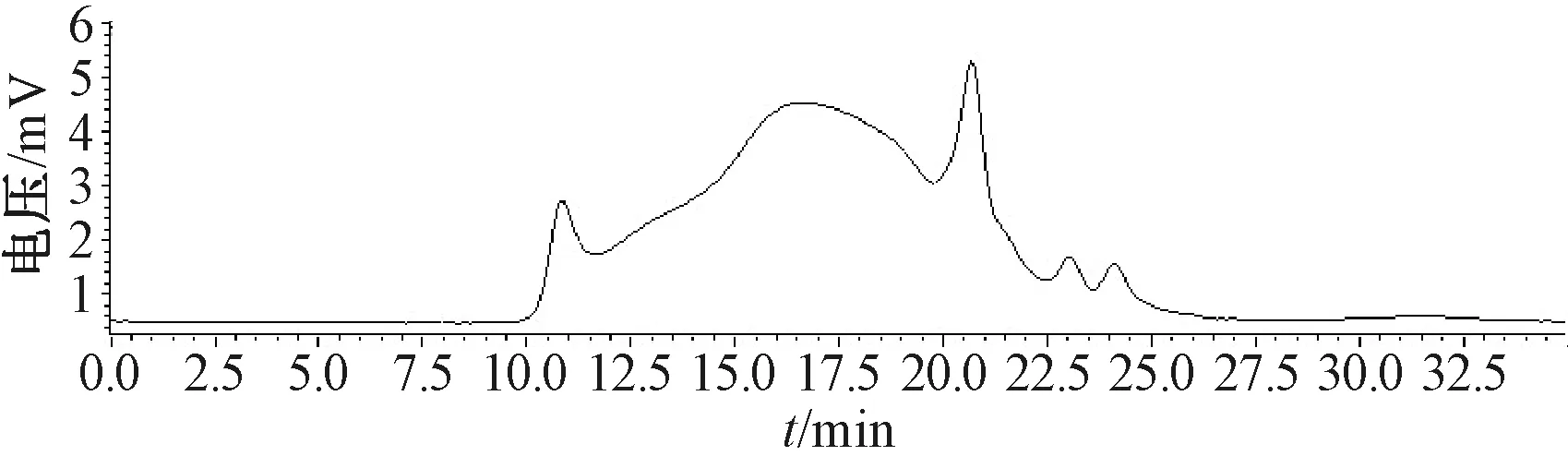
图3 单根色谱柱多糖样品HPGPC色谱图
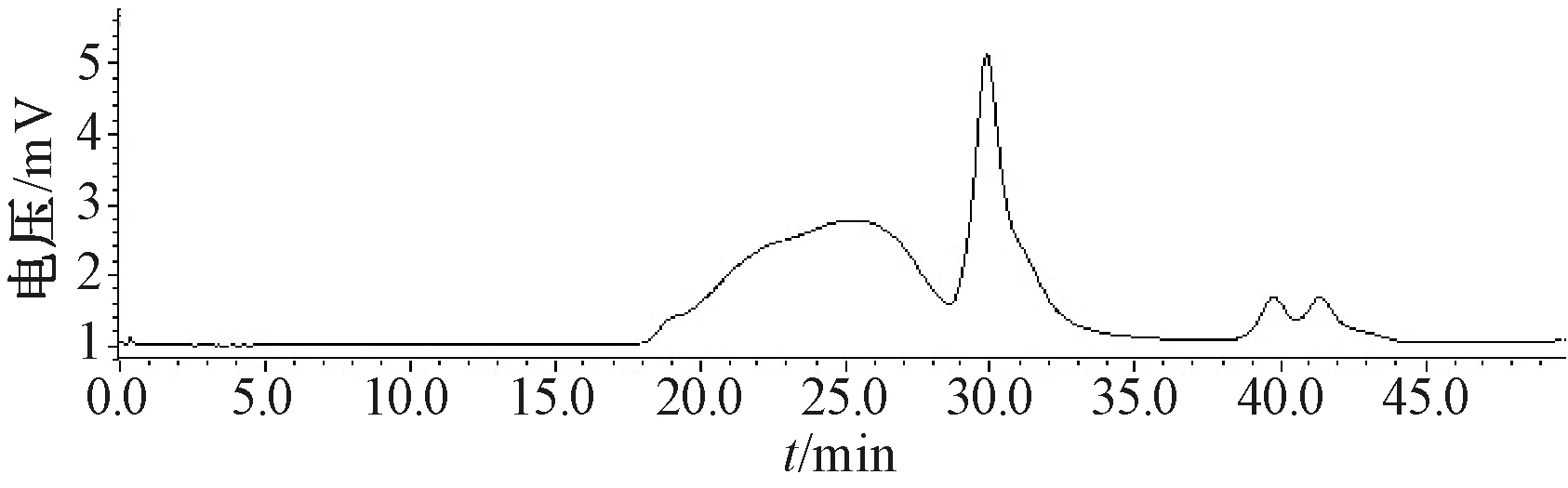
图4 串联色谱柱多糖样品HPGPC色谱图
3.2 单糖组成分析
3.2.1 多糖部位酸水解种类选择 分别采用硫酸、三氟乙酸对多糖部位进行酸水解并衍生后,采用紫外检测器测定各单糖含量,结果见表3。由此可知,三氟乙酸水解后除半乳糖醛酸含量低于硫酸水解后外,其他单糖的量均更高,而且该试剂腐蚀性小,沸点较低,容易除去,故本实验选择其对多糖部位进行水解。

表3 硫酸、三氟乙酸水解多糖后单糖含量比较
3.2.2 水解条件选择 分别以0.4 mol/L三氟乙酸、2 mol/L三氟乙酸对多糖部位水解2、4、6 h,按“2.2.3”项下方法制备衍生物,在“2.2.1”项色谱条件下进样测定,结果见表4。由此可知,0.4 mol/L三氟乙酸水解4 h、0.4 mol/L三氟乙酸水解6 h、2 mol/L三氟乙酸水解6 h得到的单糖种类相同,综合考虑成本和效率,本实验选择0.4 mol/L三氟乙酸水解4 h。
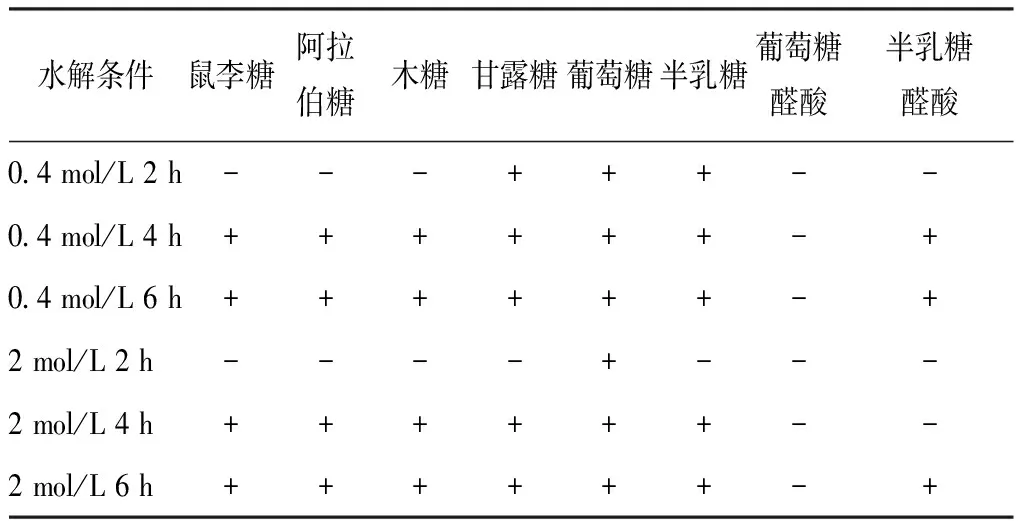
表4 水解条件考察结果
3.2.3 结果分析 多糖部位经过三氟乙酸水解后采用GC-MS进行分析,并与NIST数据库进行匹配,结合单糖标准品衍生化后比对保留时间,初步鉴定出7种单糖衍生物,色谱图见图5,GC-MS裂解碎片与NIST质谱数据库的对比见图6,由于阿拉伯糖、木糖均为五碳醛糖,甘露糖、葡萄糖、半乳糖均为六碳醛糖,衍生后质谱碎片相似,故在NIST质谱数据库中得到的结构信息也相似。表5、图5显示,多糖水解液在鼠李糖、阿拉伯糖、木糖、甘露糖、葡
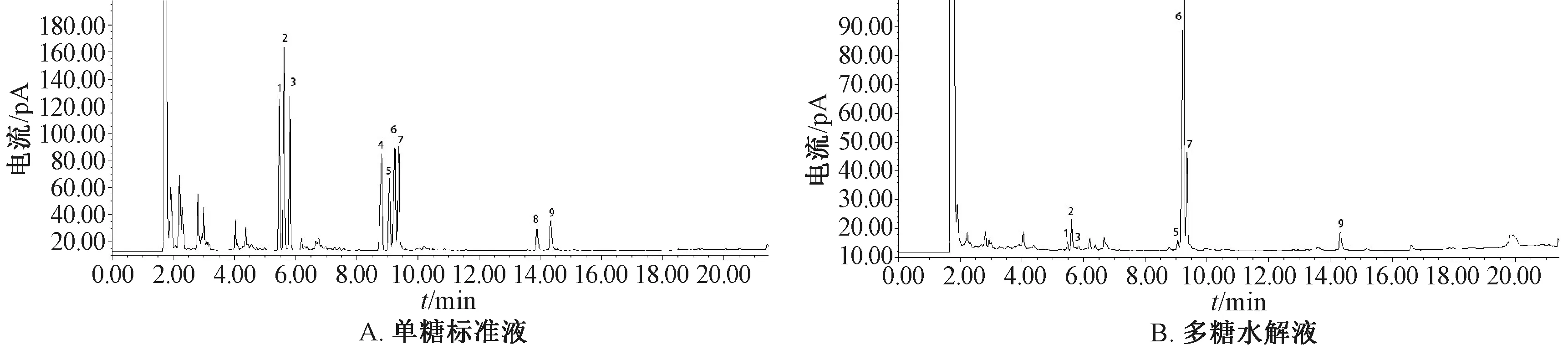
1.鼠李糖 2.阿拉伯糖 3. 木糖 4. 肌醇 5. 甘露糖 6. 葡萄糖 7. 半乳糖 8.葡萄糖醛酸 9. 半乳糖醛酸图5 各单糖GC色谱图
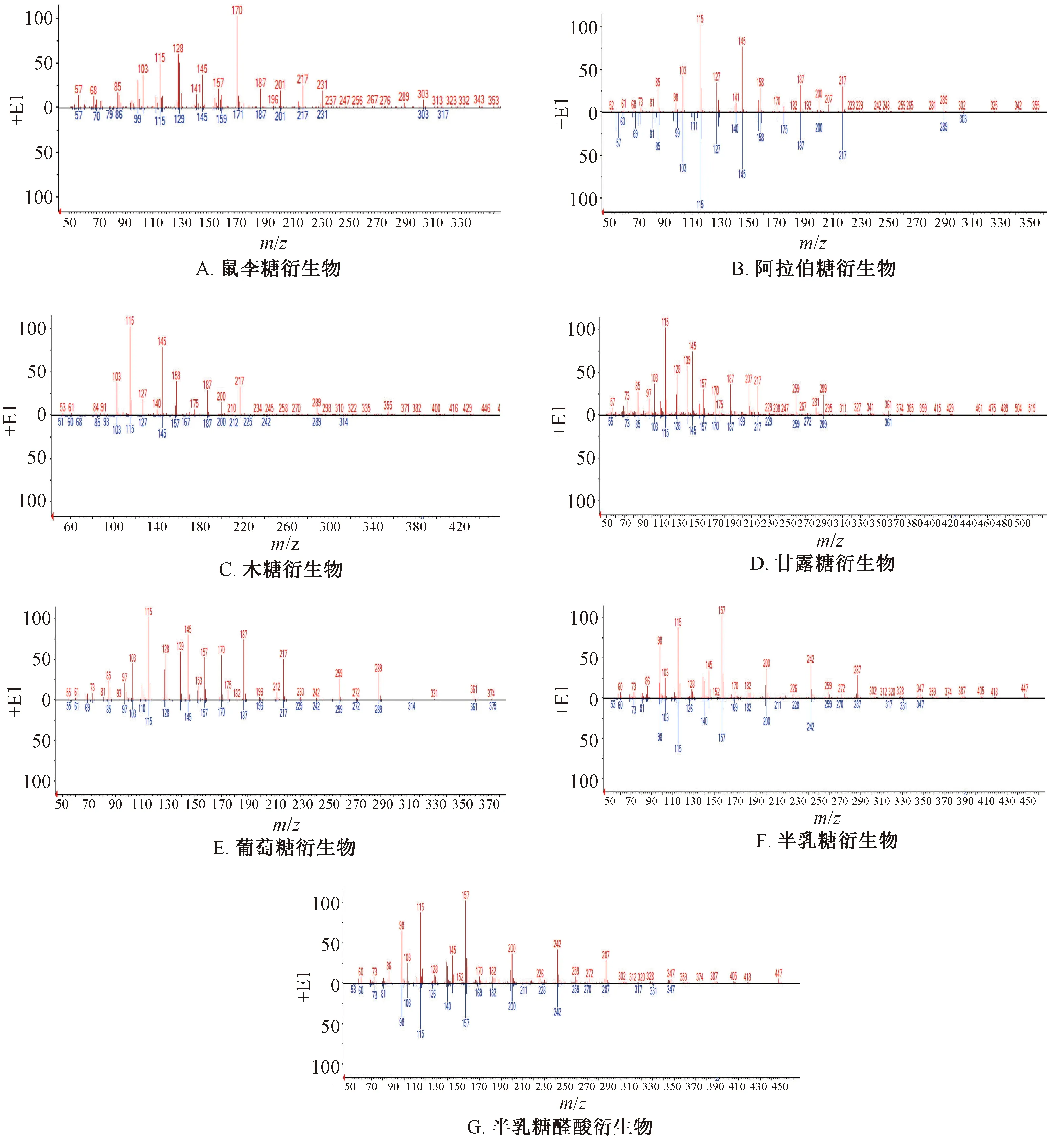
注:图中央横线上方数字为样品裂解数据,下方数字为数据库裂解数据。图6 各色谱峰GC-MS裂解碎片与NIST质谱数据库比较
萄糖、半乳糖、半乳糖醛酸7种单糖相同保留时间处均存在对应色谱峰,并且多糖水解液中葡萄糖含量最高。
任凤霞[16]等报道,六味地黄汤中多糖部位由鼠李糖、阿拉伯糖、葡萄糖、半乳糖、半乳糖醛酸组成,而未检测到木糖与甘露糖;本实验发现,多糖水解液中木糖、甘露糖含量较低,表明GC法可提高检测灵敏度。
4 讨论与结论
六味地黄苷糖片是按照由“中药饮片组方(饮片配伍)向中药复方活性成分群组方(组分配伍)发展”的中药经典方剂“二次开发”思路,多糖部位是该制剂发挥免疫调节作用的主要部位,但该成分具有难气化、无紫外吸收等特点,导致相关研究成为中药领域的难点[14-16]。本实验采用乙醇、丙酮、乙醚对六味地黄苷糖片多糖部位进行脱脂,并除去色素及小分子杂质。由于HPGPC测定多糖部位相对分子质量以Na2SO4溶液为流动相时,在固定位置有倒峰,从而影响测定,故本实验选择磷酸盐溶液作为流动相。仅以TSK-GEL G4000PWXL色谱柱为固定相时,色谱峰分离度差,而串联TSK-GEL G1000PW色谱柱后峰形和分离效果较好,最终测得其相对分子质量在10万~20万之间。再通过GC-MS法对六味地黄苷糖片多糖部位进行分析,发现它由鼠李糖、阿拉伯糖、木糖、甘露糖、葡萄糖、半乳糖、半乳糖醛酸组成,其中葡萄糖含量最高,而且结构中含有糖醛酸,推测该聚糖可能主要由葡萄糖组成,同时各批次之间单糖组成无明显差异。
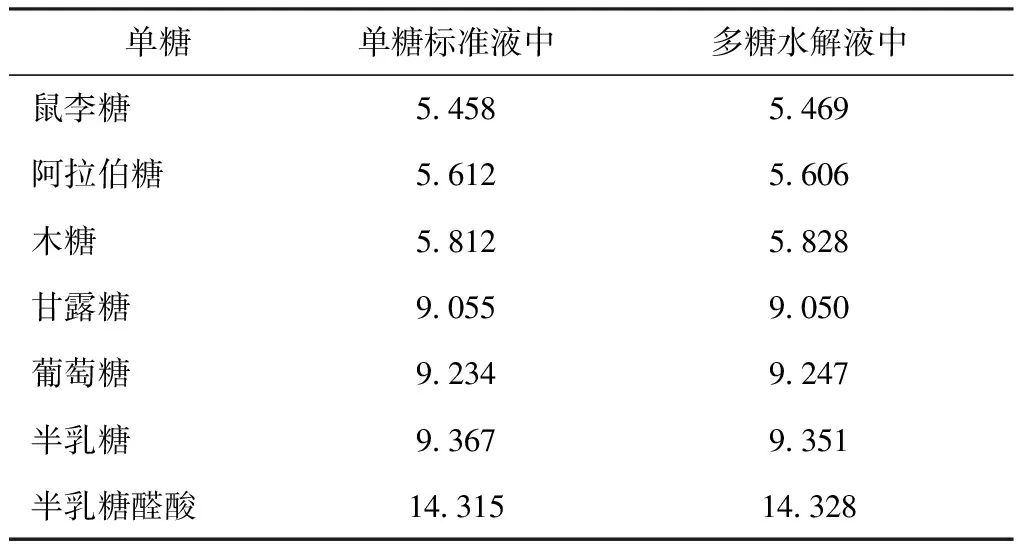
表5 单糖标准液、多糖水解液中单糖GC保留时间比较(min)
综上所述,本实验完善了六味地黄苷糖片多糖部位原料及制剂的质量标准,可为该制剂质量控制提供理论依据。
猜你喜欢
基层中医药(2022年3期)2022-07-22
中老年保健(2021年8期)2021-08-24
家庭科学·新健康(2021年5期)2021-06-21
发酵科技通讯(2021年1期)2021-03-18
家庭百事通·健康一点通(2020年12期)2020-12-31
教育教学论坛(2018年6期)2018-03-15
中国医药导报(2017年31期)2017-12-19
中国中药杂志(2017年20期)2017-11-11
科技资讯(2017年20期)2017-08-22
分析化学(2015年10期)2015-11-03
