
基于杂交种群体的玉米产量及其配合力的全基因组关联分析
2022-05-17 02:17李周帅董远李婷冯志前段迎新杨明羡徐淑兔张兴华薛吉全
中国农业科学 2022年9期
李周帅,董远,李婷,冯志前,段迎新,杨明羡,徐淑兔,张兴华,薛吉全
基于杂交种群体的玉米产量及其配合力的全基因组关联分析
李周帅,董远,李婷,冯志前,段迎新,杨明羡,徐淑兔,张兴华,薛吉全
西北农林科技大学农学院/西北旱区玉米生物与遗传改良重点实验室,陕西杨凌 712100
【】通过分析陕A群和陕B群选育自交系组配的杂交种产量,评估自交系的配合力,并开展以产量和配合力为目标性状的全基因组关联分析,挖掘产量及其配合力的关联位点,为陕A群和陕B群选育玉米自交系的改良及育种中的应用提供依据。基于NCⅡ遗传设计,以陕A群和陕B群选育的85份优良玉米自交系为亲本,构建包含246份F1的杂交种群体,在3个环境下进行产量测试,并评估产量的一般配合力和特殊配合力;利用6H90K芯片进行亲本基因型检测,获得63 879个高质量SNP标记,并进行群体遗传特征分析,在杂交种群体推测出高质量SNP标记55 951个,采用加性模型和非加性模型对杂交种产量、一般配合力和特殊配合力开展了全基因组关联分析,并基于B73参考基因组对显著关联SNPs内的基因进行挖掘和功能注释。3个环境下的产量表现符合正态分布且变异广泛,产量广义遗传力为59.04%,环境效应显著;杂交种产量、一般配合力和特殊配合力三者之间均达到极显著相关性,杂交种产量与特殊配合力的相关性(=0.95)大于与一般配合力的相关性(=0.62);陕A群与陕B群遗传特征具有一定差异,陕A群具有较高的一般配合力。全基因组关联分析分别检测到7、5和9个SNP与杂交种产量、一般配合力和特殊配合力显著相关(-log10()>3.86),其中4个SNP为杂交种产量和特殊配合力共定位,最终锚定了17个关联SNP。对不同性状关联位点的优势等位基因型分析发现,4个GCA关联SNP受加性效应控制,F1产量BLUE关联位点可分为4种表现形式,以显性效应为主,其杂合基因型为最优等位基因型或次优等位基因型。通过功能注释发现,候选基因在玉米生长发育和籽粒建成中特异表达,例如、均与玉米籽粒发育相关。一般配合力和特殊配合力共同影响杂交种的产量,特殊配合力效应影响更大;一般配合力和特殊配合力具有不同的遗传基础,可通过有利等位基因聚集提高一般配合力。在F1杂交种群体采用全基因组关联分析策略可开展配合力相关遗传解析,挖掘产量及其配合力相关遗传位点,可加速关联位点在分子育种中的应用。
玉米;杂交种;一般配合力;特殊配合力;全基因组关联分析
0 引言
【研究意义】玉米(L.)作为中国第一大粮食作物,在食品、饲料和工业原料等行业被广泛应用,在国家粮食安全体系中不可或缺[1]。当前,全球环境愈加多变复杂,为确保玉米持续增产和稳产,加速玉米种质创新,提高品种高产稳产性迫在眉睫[2]。【前人研究进展】玉米成为世界上种植面积最大、总产量最高的粮食作物,关键在于杂种优势的应用和杂交种的种植与推广。杂种优势(heterosis)是指遗传多样性个体的杂交后代在产量、适应性和抗逆性等多方面上超过双亲的现象[3-5]。以此为基础开展玉米种质改良,组配在产量、环境适应性、抗病性和品质等方面优良的品种。如堵纯信等[6]选育的郑单958和先锋公司推出的先玉335都具备高产稳产的优良特性,在中国的玉米生产中推广时间长,种植面积广,是玉米杂种优势利用的典型。长期以来,育种家使用配合力作为评价自交系和杂交种是否优良的一个重要指标[7],Sprague等[8]率先提出了配合力的概念,并将其进一步分解为一般配合力(general combining ability,GCA)和特殊配合力(special combining ability,SCA)。GCA是指某个自交系与其他自交系组配产生的杂交组合在某一农艺性状上的平均表现;SCA反映的是特定双亲组配的杂交组合在某一性状表现优于或次于双亲平均表现组合预期的部分。GCA反应自交系的育种潜力,SCA评价杂交种的实际表现和预测双亲的杂种优势。基于此对大量的种质遗传背景进行了探究:李明顺等[9]采用NCⅡ遗传设计对18份玉米自交系进行配合力分析,将其分为5个杂种优势群并鉴定出4份育种潜力高的自交系。杨爱国等[10]对27份来自CIMMYT和国内玉米群体的材料进行配合力和杂种优势分析,鉴定出9份热带、亚热带高配合力自交系,其中,Suwan1群体表现最优。除此之外,对配合力遗传基础的研究也逐步深入,Griffing[11]最早进行了配合力遗传基础的研究,将其解释为加性和非加性效应的互作并估算了GCA和SCA的遗传效应。随着分子生物学技术的发展,研究人员开始通过连锁定位和关联分析深入开展配合力遗传基础的解析工作。LÜ等[12]利用掖478及其近等系群体(含65份自交系)和3个测验种测交产生的含有198份F1的群体对产量相关性状进行QTL定位,在产量相关性状中检测到69个QTL,而产量GCA仅检测到9个QTL,表明了二者的相关遗传位点是不同的。Qi等[13]利用一个含有75份自交系的近等系群体和4个测验种组配产生的含300份F1杂交组合的群体,对多环境下产量相关性状的GCA和SCA进行了QTL定位,发现两者的相关性不显著,在GCA和SCA中分别检测到56和21个QTL,其中,只有5个同时控制GCA和SCA,说明二者的遗传基础不同,同时检测到GCA位点数与相应性状GCA表现呈显著正相关,说明GCA有利位点的累加可有效提高GCA。Wang等[14]创制了一个由724份杂交种组成的多杂交群体(multiple-hybrid population,MHP),使用TASSEL和PEPIS 2种方法对花期相关性状分别进行了全基因组关联分析,同时鉴定到5个与开花性状相关染色体区域。Zhou等[15]利用328份玉米重组自交系与2个测验种组配出656份F1杂交组合,对株高相关性状进行QTL定位,在GCA、F1和SCA分别检测到21、30和17个QTL,表明三者的遗传基础不同。Chen等[16]开发了一套NCⅡ遗传设计的384份F1构成的水稻杂交种群体对农艺性状、GCA和SCA进行关联分析,鉴定到了34个显著SNP,其中、和的优势等位基因积累对GCA表现呈正相关。Xiao等[17]利用42 820份F1组成的玉米杂交种群体,利用机器学习和全基因组关联分析等手段,解析了玉米杂种优势和配合力的遗传学基础,提出了“显性-互作”的共调控模型。全基因组关联分析在剖析复杂性状的遗传基础上取得了较大进展,如产量及产量构成因子[18]、抗旱性[19]、品质[20-21]、可变剪切[22]、可塑性[23]等,成为复杂数量性状遗传研究的一种主流技术手段。【本研究切入点】目前,对配合力的遗传基础以及利用杂交种F1代开展数量遗传学分析仍比较少,尚未有相关基因被克隆,较多研究集中在QTL定位,但由于群体多样性限制使其不能完全剖析SCA的遗传基础。【拟解决的关键问题】本研究基于NCII遗传设计以陕A群和陕B群选育的85份玉米自交系为亲本,组配了246份F1杂交组合,在3个环境下对参试材料进行配合力评价和鉴定,并对产量及其配合力进行全基因组关联分析,挖掘与产量、产量GCA和SCA显著相关的SNP,最后结合功能注释预测相应的基因,以期挖掘产量杂种优势位点进行功能分析并开发相应的标记应用于玉米分子辅助设计育种。
1 材料与方法
1.1 试验材料与田间设计
所涉及的85份玉米自交系均来源于西北旱区玉米生物学与遗传育种重点实验室自主创建的陕A群和陕B群选育的玉米自交系。采用NCII遗传设计,于2017年冬季在西北农林科技大学海南三亚南繁基地(109.20°E,18.40°N)进行杂交测配,获得246份F1杂交组合。具体为:1)以陕A群的KA064、KA105和91227为测验种与陕B群的61份自交系为被测系进行组配产生183份F1杂交组合;2)以陕B群的KB106、KB207和KB588作为测验种与陕A群的21份自交系为被测系进行组配产生63份F1杂交组合(电子附表1)。246份F1杂交组合分别于2018年4月在榆林(109.78°E,38.24°N)和旬邑(108.33°E,35.11°N)、6月在杨凌(108.05°E,34.31°N)3个环境下种植,采用α-格子设计,2次重复,密度为90 000株/hm2,每小区4行,行长5 m,行距0.6 m。各环境田间管理措施同当地大田生产管理一致。
1.2 田间产量性状调查与数据分析
成熟期收获小区中间两行玉米果穗,称量总鲜重记为W,总穗数为N,计算平均单穗鲜重W/N,根据平均单穗鲜重挑选出10个均匀果穗,自然晾晒后脱粒,称量籽粒干重,用PM-8188谷物水分测定仪测定玉米籽粒的含水量,折合成14%标准收获含水量后计算小区产量,用kg·hm-2表示。产量(kg hm-2)=单穗籽粒干重(kg)×收获行有效株数×(1-籽粒含水量)/(1-14%)/两行小区面积(m2)×10000。利用QTL IciMapping[24]和R语言(https://cran.r-project.org/)对原始数据进行描述性统计、相关性分析、方差分析。广义遗传力(broad-sense heritability,2)按照Knapp等[25]提出的公式2=g2/(σ2+σ2/+σ2/)计算,其中,σ2为遗传方差,σ2为基因型与环境互作方差,σ2为误差方差,为环境数,为重复。使用最佳线性无偏估计值(best linear unbiased estimator,BLUE)作为F1产量性状的表型值,以最大降低环境等因素对每个材料产量造成的偏差。同时用R包sommer[26]对246个F1杂交组合的产量SCA以及被测系的产量GCA进行计算,GCA和SCA的计算公式[27]分别为:GCA=-;SCA=-GCA--,其中为自交系组配所有F1的表型均值,为自交系与自交系组配的F1的表型值,GCA为亲本的GCA,GCA为亲本的GCA,为所有F1的表型均值,并同时对配合力进行了相关性分析。本研究对82份被测系的产量GCA(统称为被测系GCA)、246份F1杂交组合的产量BLUE值(统称为F1产量BLUE)和SCA(统称为SCA)进行后续分析。
1.3 基因型提取检测与SNP质量控制
每份自交系随机挑取10株五叶期幼嫩叶片,-80℃保存,用于DNA提取与基因型检测。采用改良的CTAB法[28]提取DNA,之后通过琼脂糖凝胶电泳、凝胶成像仪、紫外分光光度计(Nanodrop2000,Thermo Scientific)等检测仪器和技术进行DNA质量和浓度测定,检测合格后使用Affymetrix maize 6H90K芯片(北京康普森生物技术有限公司)进行基因分型。被测系使用基因型为以缺失率>0.2,最小等位基因频率(minor allele frequency,MAF)<0.05的标准质控后获得的63 879个高质量SNP。杂交种基因型由双亲基因型推算获得,具体方法为:将亲本中杂合基因位点均记为缺失并使用VCFtools工具[29]剔除缺失率>0.2的SNPs后使用Beagle软件[30]进行填补,基于双亲基因型对F1基因型进行推算,对推算所得SNPs按照MAF<0.05,缺失率>0.2进行过滤,最终获得55 951个高质量SNP。
1.4 聚类分析和主成分分析
基于MEGA-X[31]中的NJ(neighbor joining)法[32]构建亲本发育树,并在iTOL[33]中进行可视化。亲本和F1群体的主成分分析通过GCTA[34]进行计算,在R语言中可视化。
1.5 全基因组关联分析
使用R包GAPIT[35]中的BLINK方法[36]对被测系GCA、F1产量BLUE和SCA进行全基因组关联分析。由于F1群体的基因型高度杂合,受加性和非加性效应共同影响,基因型的加性编码不能反映非加性效应,根据Huang等[37]基因型编码方式对F1群体的基因型进行重新编码,具体为(1)显性编码方式:将杂合等位基因型改为主等位基因型;(2)隐性编码方式:将杂合等位基因型改为次等位基因型,并将2种编码方式统一表示为显性模型。后分别进行加性、非加性F1相关性状的全基因组关联分析。为了控制假阳性,根据Gao等[38]提出的方法对有效标记数(N)进行计算得到720个有效标记,并按照阈值=-log10(0.1/N)的标准将阈值确定为3.86,以此筛选显著SNP,同时对显著SNP按照2>0.6的标准筛选连锁位点,保留其中值较小的SNP。以显著SNP在玉米B73参考基因组(ZmB73_RefGen_v3; http:// www.maizeGDB.org/)中的物理位置最近的基因为候选基因,进行功能注释。
2 结果
2.1 产量统计分析及方差分析
通过对同一环境下2个重复间的产量进行相关性分析,结果显示,3个环境下的两重复间产量数据均达到极显著相关水平(榆林=0.74**、旬邑=0.76**、杨凌=0.38**),杨凌点重复区组效应较大,远大于榆林和旬邑。同时,夏播的杨凌点产量均值(9 407.47 kg·hm-2)显著低于春播点的榆林(15 663.80kg·hm-2)和旬邑(14 076.95kg·hm-2),且杨凌2个重复的变异幅度(13%和15%)均较大。在3个环境下,群体产量的变异幅度均大于5%(表1),说明试验群体的产量在不同环境下均具有广泛的变异;偏度和峰度的绝对值均处于0—1,说明不同环境下不同材料的产量分布均服从正态分布,且为一个典型的数量性状。方差分析表明基因型、环境、基因型环境互作3种效应均达到了极显著水平,F1群体的广义遗传力为59.04%,说明不同材料间的遗传背景差异显著,产量性状受环境效应影响极大,且3个环境的效应差异显著;产量性状受遗传效应、环境效应和基因环境互作效应共同调控。因此,为了更好地进行后续的全基因组关联,使用BLUE值作为表型,最大程度上降低环境效应对遗传效应的影响。
2.2 配合力评价
被测系GCA和杂交种SCA统计分析(表2)可知SCA效应范围(-1 772.11—1 621.62)远大于GCA效应变化范围(-565.87—316.87),其受非加性效应影响。GCA和SCA的偏度和峰度绝对值均小于1(表2),说明GCA和SCA的分布符合正态分布。对GCA、SCA和F1群体的产量BLUE值进行相关性分析(表2),结果表明,GCA、SCA和产量三者之间均呈现极显著相关,其中,GCA和SCA的相关系数(=0.51)最低,说明二者作用方式存在差异。不同测验种与其组配的F1杂交组合产量的比较(图1)表明,不同测验种表现出的杂种优势有所差异,如测验种KA105具有较高产量,其组配F1杂交组合的产量也在群体中处于较高水平,而测验种KB106及其子代产量均处于较低水平,说明高产自交系更容易组配出高产组合。

表1 杂交种群体产量描述统计分析和方差分析
*:在<0.05水平差异显著;**:在<0.01水平差异极显著。BLUE:最佳线性无偏估计。SD:标准偏差。CV:变异系数。下同
*: significance at<0.05; **: significance at<0.01. BLUE: Best linear unbiased estimator; SD: Standard deviation; CV: Coefficient of variation. The same as below
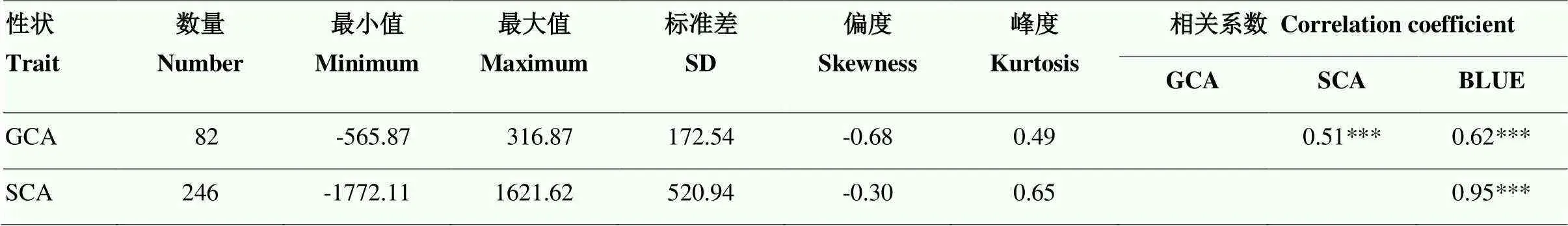
表2 配合力基本统计分析
GCA:一般配合力;SCA:特殊配合力;***:在<0.001水平差异显著。下同
GCA: general combining ability; SCA: special combining ability; ***: significance at<0.001. The same as below

图1 测验种及其组配F1杂交组合产量比较分析
测验种KA105和KB588的产量和及其组配的F1杂交组合的产量高于同组其他2个测验种,说明KA105和KB588具有更优的产量遗传背景。按照自交系类群,对82份被测系的GCA进行评价(图2)发现,具有正向GCA效应的自交系共46份,占被测系总体的56%,其中,源于陕A群被测系16份,约占陕A群总体的76.19%;源于陕B群被测系30份,约占陕B群总体的49.18%。在整体上,陕A群被测系的GCA也呈现高于陕B群的趋势(图2),说明陕A群选育自交系具有GCA高的遗传背景。

图2 源于陕A群、陕B群的82个被测系GCA评价
2.3 群体遗传特征分析
利用高通量SNPs标记对85份亲本进行聚类分析(图3-A),结果显示,源于陕A群选育的自交系与源于陕B群选育的自交系可以较好地区分开,总体分为3类,即一类源自陕A群的亲本;一类为源自陕B群选育的自交系;另一类为源自陕A群和陕B群选育自交系的混合群。主成分分析表明(图3-B),陕A群和陕B群选育自交系可以大致的分为2组,陕A群选育自交系位于第一象限且分布较为集中,陕B群选育自交系群体更大分布范围广,总体上向2个方向散布于二、三、四象限,该结果再次印证了源自陕B群的材料群体大,遗传背景更为丰富。同时测验种在亲本遗传背景的分布较为均衡(图3-A中测验种被标注为“T”;图3-B中测验种用黑色星号标出),说明本研究测验种选择合理,通过陕A群和陕B群之间的组配一定程度上降低了群体结构对全基因组关联分析的影响。
2.4 产量及其配合力的全基因组关联分析
基于BLINK对被测系GCA、F1产量BLUE值和SCA进行全基因组关联分析(图4)。由于该方法遵循加性编码的基因型编码方式,而产量以及SCA存在非加性效应,基因型加性编码无法定位到非加性效应的位点,因此,本研究在F1群体中同时开展了对基因型加性编码(additive,ADD)和非加性编码(dominant,DOM)的全基因组关联分析,并按照阈值-log10()>3.86对显著SNPs进行筛选。当使用加性编码基因型进行F1全基因关联分析时,在F1产量BLUE和SCA中各定位到1个显著SNP,解释表型变异率分别为1.55%和1.77%。当使用非加性编码基因型进行全基因关联分析时,在F1产量BLUE和产量SCA中的显著SNP分别为6和8个。非加性模型较加性模型检测到的显著SNP多,表明在加性模型下某些非加性效应会被掩盖而难以体现,以非加性进行基因型编码可以检测到非加性效应位点。同时以82个被测系的产量GCA为目标性状开展全基因组关联分析,共检测到5个显著SNP。除此之外,加性模型下得到的Affx-291424805,非加性模型下的Affx-291385286、Affx-88980445和Affx-291431456,4个显著SNP在产量BLUE和SCA均被定位(表3)。
从显著SNPs分布来看,被测系GCA相关的显著SNPs较少且集中于第4染色体,但表型解释率较大(7.97%—23.37%);F1产量BLUE和SCA挖掘得到的显著SNP多数集中于第2和第10染色体,数目多但贡献率较低(0.43%—6.31%)。被测系GCA和杂交种SCA不存在共定位的结果,说明被测系GCA与F1产量和SCA的遗传基础是不同的,加性效应决定了GCA表现,而SCA更多的是由非加性效应的微效多基因共同作用。

A:聚类分析;B:主成分分析 A: Clustering analysis; B: Principal component analysis

A:被测系GCA;B:基于加性模型的F1产量BLUE;C:基于显性模型的F1产量BLUE;D:基于加性模型的杂交种SCA;E:基于显性模型的杂交种SCA

表3 产量及其配合力显著关联的SNP信息
●、■、▲、★均表示共定位 ●, ■, ▲, ★ all represent the same SNPs
2.5 显著SNP分析及候选基因预测
对不同性状关联位点的优势等位基因型分析发现(表4和图5),4个GCA关联的SNP(SNP_1、SNP_2、SNP_3和SNP_5),其GCA的有利等位基因在F1产量BLUE和SCA中仍然表现为纯合基因型,说明这些位点可能完全或主要受加性效应控制;与GCA关联的SNP_4在被测系GCA中表现为AA>GG,在F1产量BLUE和SCA中杂合位点GCA与纯合位点GG无显著差异,说明该位点主要受加性控制。随着有利等位基因数量的增加,被测系GCA也呈现上升趋势,说明可以通过有利等位基因的聚合提高自交系的GCA。
F1产量BLUE关联位点在性状的表现形式分为4种;1)纯合基因型的表型无差异,杂合基因型为最优基因型,且只与低值纯合基因型差异显著,包括(SNP_6、SNP_8、SNP_9、SNP_14和SNP_16);2)纯合基因型的表型无差异,杂合基因型为最优基因型,且只与2个纯合基因型均表现为差异显著,包括(SNP_12和SNP_15);3)纯合基因型的表型无差异,杂合基因型为最差基因型,且只与高值纯合基因型差异显著,包括(SNP_7、SNP_10和SNP_11);4)纯合基因型的表型有差异,同时杂合基因型与高值纯合基因型的表型无差异,与低值基因型的表型存在显著差异(SNP_17)。由此可知,所关联的大部分位点的杂合基因型的表型与某1个或者2个纯合位点存在表型差异,说明F1产量BLUE关联的位点以显性效应为主,而不同基因型的表型分析也说明存在一定的加性效应。GCA检测的位点与F1产量和SCA无共定位,这说明产量GCA的遗传基础不同于F1产量BLUE和SCA,更可能是由加性效应为主效应的一个性状。通过对GCA显著关联位点的等位基因型的累加效应分析发现(图6),对产量及其GCA和SCA显著关联的SNP,结合maizeGDB数据库(http://www.maizeGDB.org/)以关联位点最近的基因为候选基因进行筛选,共得到17个相关的候选基因并对其进行了功能注释(表5)。


表4 显著位点效应分析
CO_ADD和CO_DOM分别表示基于加性和显性模型在F1和SCA中获得的共定位结果;D:显性效应;A:加性效应
CO_ADD and CO_DOM respectively indicate a co-SNP based on additive and dominant models in F1and SCA; D: dominant effect; A: additive effect
3 讨论
3.1 产量及其配合力遗传基础解析
配合力在育种实践中的重要作用使得解析配合力的遗传基础必不可少,本研究通过对配合力的分析,发现GCA和SCA的相关性较低,在关联分析中两者也未检测到相同区域SNP,表明控制GCA与SCA的遗传位点可能是不同的,这与Qi等[13]研究结果是一致的。对于产量性状和配合力的关系,Zhou等[39]研究显示GCA和SCA都对F1表型具有一定贡献。本研究中F1产量与其SCA相关性较高,且2个性状间存在4个共定位,分别占F1产量和SCA显著SNP总数的57.14%和44.44%,印证了其研究结论,表明二者遗传基础存在一定相似性。当使用加性模型对产量及其配合力进行关联分析时,GCA得到5个显著SNP且贡献率较大(23.37%—7.97%),F1产量和SCA仅各定位到1个SNP;而在显性模型中F1产量和SCA各有6个、8个显著SNP被检测到,说明了加性模型并不适用于杂交种的关联分析,这与Chen等[16]研究结果类似。同时也说明产量SCA和GCA遗传效应的不同,SCA以非加性效应为主,而GCA以加性效应为主。

图6 被测系GCA有利等位基因富集效应分析

表5 候选基因预测
超显性假说[40]认为杂种优势是由于杂合等位基因间的相互作用大于纯合等位基因间的相互作用而形成的,杂种优势归因于双亲的基因异质性,与等位基因的显隐性无关。梁文科等[41]认为GCA主要遵循加性遗传,SCA遗传基础以非加性效应中的显性效应为主。本研究对显著SNP结合其在不同性状的有利基因进行了区分,将在F1中表现为杂合优势的标为显性效应(D);表现和GCA相同的标为加性效应(A)。本研究发现SCA显著SNP绝大多数(88.89%)表现出杂合优势,F1产量显著SNP有57.14%表现为杂合优势,GCA显著SNP几乎不表现杂合优势,说明SCA和杂种优势有一定的紧密关系,但又有所区别[42-44]。同时通过GCA显著SNP进行有利等位基因聚合,证实亲本自交系所含的有利基因位点数和其GCA呈显著正相关,这部分效应可以稳定遗传给后代,即育种家可通过有利等位基因的累积选育GCA高的玉米自交系。
3.2 全基因组关联分析在杂交种群体中的应用
随着数量遗传学的不断发展,研究人员创制了不同植物的各种双亲、多亲以及自然群体应用于数量性状的基因挖掘。在玉米中常见的农艺性状多数为复杂的数量性状,由不同自交系组成的群体正在使用全基因组关联分析对该类性状的遗传基础进行解析。但实际上,基于自交系的全基因组关联分析并不能直接应用于剖析F1群体的遗传基础,而玉米生产中应用的却是杂交种,对杂交种的研究能更直观地解决实际问题。杂交种进行全基因组关联分析在水稻研究中应用较多,Huang等[37]使用了1 495份杂交水稻品种及其自交系构建了高密度的基因组图谱,通过全基因组关联分析检测了38个农艺性状的130个显著SNP。小麦中,Jiang等[45]在11个环境下对1 604份冬小麦杂交种及其135份亲本的产量进行了全基因组关联分析,表明了上位性效应在小麦产量杂种优势中的作用高于显性效应。玉米中,Romero等[46]基于全基因组关联分析对4 471份玉米自交系和3 552份F1群体的开花性状以及可塑性进行研究。本研究利用85份自交系通过NCⅡ遗传设计产生了246份杂交组合组成的F1群体被用于全基因组关联分析。与传统的自交系创制群体相比,F1群体的环境适应性更强,提高了多环境测试的可行性。同时在进行基因型测序时,由于只有亲本自交系可以进行基因分型,杂种子代的基因型是通过双亲基因型进行推断,因此,使用F1群体可以降低基因分型的成本。目前,扩大群体规模成为提高检测精度的重要手段,F1群体的应用可以极大简化大规模群体创制流程。但是当前全基因组关联分析主流的软件算法多数仅考虑加性效应且适用于纯合群体的分析,杂交种群体并不适用,本研究中使用加性模型对F1群体定位仅得到较少的位点,而显性模型更加适合杂交种群体分析,未来开发适用于杂交种群体的全基因组关联分析并能同时分析加性、显性、上位性效应的新研究手段是有必要的。
3.3 候选基因分析
本研究对17个候选基因结合其在玉米不同器官表达的特异性以及前人研究进行了分析。其中在GCA的SNP_3区间中检测到的(),拟南芥中的同源基因在侧生器官发育中特异表达,并调控下游影响远轴发育的转录因子[47-48]。SNP_5区间内的在玉米各个器官的表达量均较高[49-50],表明该基因对玉米各个生育阶段均十分重要,同时它与玉米雄穗分支数显著关联[51]。产量和产量SCA共定位的结果中,位于SNP_6区间,其编码木葡聚糖醛酸β-1,3-木糖基转移酶,被认为和玉米株高相关[52];位于SNP_8区间内,根据表达谱显示它在未授粉的雌穗和正常发育的胚中高表达[49-50],并且其水稻同源基因编码染色体浓缩调节蛋白。在与产量性状显著关联的SNP_12区间内定位到了候选基因,其与玉米果穗发育和根长有关[53-54]。在SCA关联的SNP_13区间内检测到的()参与玉米光周期反应,与花期相关生理过程密切相关[55];SNP_16区间候选基因在授粉后生殖生长阶段的胚中特异性表达[49-50];SNP_17区间内的被认为是一个与粒宽相关的基因,与玉米籽粒建成密切相关[56]。
4 结论
陕A群和陕B群选育玉米自交系组配F1群体的GCA和SCA共同影响杂交种的产量,且SCA的效应对杂交种的产量影响要大于GCA,育种中作为母本的陕A群选育玉米自交系具有更高的GCA。杂交种产量受加性和非加性效应共同影响,非加性模型对检测杂交种位点具有更高的效率;同时GCA和SCA可能具有不同的遗传基础,GCA受加性效应主导可稳定遗传,且随其有利等位基因的富集而提高,SCA主要受非加性效应影响。产量及其配合力相关的候选基因在玉米生长发育和籽粒建成中特异性表达。
[1] 李少昆, 赵久然, 董树亭, 赵明, 李潮海, 崔彦宏, 刘永红, 高聚林, 薛吉全, 王立春, 王璞, 陆卫平, 王俊河, 杨祁峰, 王子明. 中国玉米栽培研究进展与展望. 中国农业科学, 2017, 50(11): 1941-1959.
LI S K, ZHANG J R, DONG S T, ZHAO M, LI C H, CUI Y H, LIU Y H, GAO J L, XUE J Q, WANG L C, WANG P, LU W P, WANG J H, YANG Q F, WANG Z M. Advances and prospects of maize cultivation in China. Scientia Agricultura Sinica, 2017, 50(11): 1941-1959. (in Chinese)
[2] 李鹏, 白永新, 张润生, 魏振飞, 张建华. 浅议我国玉米育种发展现状与方向. 种子科技, 2019, 37(2): 18-19.
LI P, BAI Y X, ZHANG R S, WEI Z F, ZHANG J H. Discussion on the development status and direction of maize breeding in China. Seed science and technology, 2019, 37(2): 18-19. (in Chinese)
[3] Birchler J A, Auger D L, Riddle N C. In search of the molecular basis of heterosis. The Plant cell, 2003, 15(10): 2236-2239.
[4] Birchler J A, Yao H, Chudalayandi S. Unraveling the genetic basis of hybrid vigor. Proceedings of the National Academy of Sciences of the USA, 2006, 103(35): 12957.
[5] Birchler J A, Yao H, Chudalayandi S, Vaiman D, Veitia R A. Heterosis. The Plant cell, 2010, 22(7): 2105-2112.
[6] 堵纯信, 曹春景, 曹青, 毕蒙蒙, 董战鲲, 张发林. 玉米杂交种郑单958的选育与应用. 玉米科学, 2006(6): 43-45+49.
DU C X, CAO C J, CAO Q, BI M M, DONG Z K, ZHANG F L. The breeding and application of maize hybrid Zhengdan 958. Journal of Maize Sciences, 2006(6): 43-45+49. (in Chinese)
[7] 钏兴宽. 配合力理论及其在水稻育种中的应用. 种子, 2014, 33(6): 39-41+46.
CHUAN X K. Combining ability theory and its application in rice breeding. Seed, 2014, 33(6): 39-41+46. (in Chinese)
[8] Sprague G, Tatum L. General vs. specific combining ability in single crosses of corn. Agronomy Journal, 1942, 34: 923-932.
[9] 李明顺, 张世煌, 李新海, 潘光堂, 白丽, 彭泽斌. 根据产量特殊配合力分析玉米自交系杂种优势群. 中国农业科学, 2002, 35(6): 600-605.
LI M S, ZHANG S H, LI X H, PAN G T, BAI L, PENG Z B. Study on heterotic groups among maize inbred lines based on SCA. Scientia Agricultura Sinica, 2002, 35(6): 600-605. (in Chinese)
[10] 杨爱国, 张世煌, 李明顺, 荣廷昭, 潘光堂. CIMMYT和我国玉米种质群体的配合力及杂种优势分析. 作物学报, 2006, 32(9): 1329-1337.
YANG A G, ZHANG S H, LI M S, RONG T Z, PAN G T. Combining ability and heterosis of 14 CIMMYT and 13 domestic maize populations in an NCⅡ mating design. Acta Agronomica Sinica, 2006, 32(9): 1329-1337. (in Chinese)
[11] Griffing B. Concept of general and specific combining ability in relation to diallel crossing systems. Australian Journal of Biological Sciences, 1955, 9: 463-493.
[12] LÜ A Z, Zhang H, Zhang Z X, Tao Y S, Yue B, Zheng Y L. Conversion of the statistical combining ability into a genetic concept. Journal of Integrative Agriculture, 2012, 11(1): 43-52.
[13] Qi H, Huang J, Zheng Q, Huang Y, Shao R, Zhu L, Zhang Z, Qiu F, Zhou G, Zheng Y, Yue B. Identification of combining ability loci for five yield-related traits in maize using a set of testcrosses with introgression lines. Theoretical and Applied Genetics, 2013, 126(2): 369-377.
[14] Wang H, Xu C, Liu X, Guo Z, Xu X, Wang S, Xie C, Li W X, Zou C, Xu Y. Development of a multiple-hybrid population for genome-wide association studies: Theoretical consideration and genetic mapping of flowering traits in maize. Scientific Reports, 2017, 7(1): 40239.
[15] Zhou Z, Zhang C, Lu X, Wang L, Hao Z, Li M, Zhang D, Yong H, Zhu H, Weng J, Li X. Dissecting the genetic basis underlying combining ability of plant height related traits in maize. Frontiers in plant science, 2018, 9: 1117.
[16] Chen J, Zhou H, Xie W, Xia D, Gao G, Zhang Q, Wang G, Lian X, Xiao J, He Y Q. Genome-wide association analyses reveal the genetic basis of combining ability in rice. Plant Biotechnology Journal, 2019, 17(11): 2211-2222.
[17] Xiao Y, Jiang S, Cheng Q, Wang X, Yan J, Zhang R, Qiao F, Ma C, Luo J, Li W, Liu H, Yang W, Song W, Meng Y, Warburton M, Zhao J, Wang X, Yan J. The genetic mechanism of heterosis utilization in maize improvement. Genome Biology, 2021, 22(1): 148.
[18] Huang X, Yang S, Gong J, Zhao Q, Feng Q, Zhan Q, Zhao Y, Li W, Cheng B, Xia J, Chen N, Huang T, Zhang L, Fan D, Chen J, Zhou C, Lu Y, Weng Q, Han B. Genomic architecture of heterosis for yield traits in rice. Nature, 2016, 537(7622): 629-633.
[19] Wang H, Qin F. Genome-wide association study reveals natural variations contributing to drought resistance in crops. Frontiers in Plant Science, 2017, 8: 1110.
[20] Li H, Peng Z, Yang X, Wang W, Fu J, Wang J, Han Y, Chai Y, Guo T, Yang N, Liu J, Warburton M L, Cheng Y, Hao X, Zhang P, Zhao J, Liu Y, Wang G, Li J, Yan J. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nature Genetics, 2013, 45(1): 43-50.
[21] Li X, Wang M, Zhang R, Fang H, Fu X, Yang X, Li J. Genetic architecture of embryo size and related traits in maize. The Crop Journal, 2022, 10(1): 204-215.
[22] Chen Q, Han Y, Liu H, Wang X, Sun J, Zhao B, Li W, Tian J, Liang Y, Yan J, Yang X, Tian F. Genome-wide association analyses reveal the importance of alternative splicing in diversifying gene function and regulating phenotypic variation in maize. The Plant Cell, 2018, 30(7): 1404-1423.
[23] Liu N, Du Y, Warburton M, Xiao Y, Yan J. Phenotypic plasticity contributes to maize adaptation and heterosis. Molecular Biology and Evolution, 2020, 38(4): 1262-1275.
[24] Meng L, Li H, Zhang L, Wang J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. The Crop Journal, 2015, 3(3): 269-283.
[25] Knapp S J, Stroup W W, Ross W M. Exact confidence intervals for heritability on a progeny mean basis. Crop Science,1985, 25(1): 192-194.
[26] Covarrubias-Pazaran G. Genome-assisted prediction of quantitative traits using the R package sommer. PloS one, 2016: e0156744.
[27] 黄远樟, 刘来福. 作物数量遗传学基础: 六、配合力: 不完全双列杂交. 遗传, 1980(2): 43-46.
HUANG Y Z, LIU L F. Basis of crop quantitative genetics: VI. combining ability: incomplete diallel hybridization. Genetics, 1980(2): 43-46. (in Chinese)
[28] Murray M G, Thompson W F. Rapid isolation of high molecular weight plant DNA. Nucleic acids research, 1980, 8(19): 4321-4325.
[29] Danecek P, Auton A, Abecasis G, Albers C A, Banks E, Depristo M A, Handsaker R E, Lunter G, Marth G T, Sherry S T, Mcvean G, Durbin R. 1000 genomes project analysis group. The variant call format and VCFtools. Bioinformatics (Oxford, England), 2011, 27(15): 2156-2158.
[30] Ayres D L, Darling A, Zwickl D J, Beerli P, Holder M T, Lewis P O, Huelsenbeck J P, Ronquist F, Swofford D L, Cummings M P, Rambaut A, Suchard M A. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Systematic biology, 2012, 61(1): 170-173.
[31] Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular biology and evolution, 2018, 35(6): 1547-1549.
[32] Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 1987, 4(4): 406-425.
[33] Letunic I, Bork P. Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics, 2007, 23(1): 127-128.
[34] Yang J, Lee S H, Goddard M E, Visscher P M. GCTA: a tool for genome-wide complex trait analysis. American journal of human genetics, 2011, 88(1): 76-82.
[35] Lipka A E, Tian F, Wang Q, Peiffer J, Li M, Bradbury P J, Gore M A, Buckler E S, Zhang Z. GAPIT: genome association and prediction integrated tool. Bioinformatics (Oxford, England), 2012, 28(18): 2397-2399.
[36] Huang M, Liu X, Zhou Y, Summers R M, Zhang Z. BLINK: a package for the next level of genome-wide association studies with both individuals and markers in the millions. Giga Science, 2019, 8(2): 154.
[37] Huang X, Yang S, Gong J, Zhao Y, Feng Q, Gong H, Li W, Zhan Q, Cheng B, Xia J, Chen N, Hao Z, Liu K, Zhu C, Huang T, Zhao Q, Zhang L, Fan D, Zhou C, Lu Y, Weng Q, Wang Z X, Li J, Han B. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nature Communications, 2015, 6(1): 6258.
[38] Gao X, Starmer J, Martin E R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genetic Epidemiology, 2008, 32(4): 361-369.
[39] Zhou H, Xia D, Zeng J, Jiang G, He Y. Dissecting combining ability effect in a rice NCII-III population provides insights into heterosis in indica-japonica cross. Rice, 2017, 10(1): 39.
[40] Shull G H. The composition of a field of maize. Journal of Heredity, 1908, 4(1): 296-301.
[41] 梁文科, 张世煌, 戚廷香, 邱法展, 庹洪章, 刘永忠, 郑用琏, 徐尚忠. 热带温带玉米群体产量性状遗传力及遗传方差分量的剖析. 中国农业科学, 2006, 39(11): 2178-2185.
LIANG W K, ZHANG S H, QI T X, QIU F Z, TUO H Z, LIU Y Z, ZHENG Y L, XU S Z. Dissection of heritability and genetic variance components for yield traits in tropical and temperate maize populations. Scientia Agricultura Sinica, 2006, 39(11): 2178-2185. (in Chinese)
[42] Makumbi D, Betrán J F, Bänziger M, Ribaut J M. Combining ability, heterosis and genetic diversity in tropical maize (L.) under stress and non-stress conditions. Euphytica, 2011, 180(2): 143-162.
[43] Zhang X, Lv L, Lv C, Guo B, Xu R. Combining ability of different agronomic traits and yield components in hybrid barley. PloS one, 2015: e0126828.
[44] 倪先林, 张涛, 蒋开锋, 杨莉, 杨乾华, 曹应江, 文春阳, 郑家奎. 杂交稻特殊配合力与杂种优势、亲本间遗传距离的相关性. 遗传, 2009, 31(8): 849-854.
NI X L, ZHANG T, JIANG K F, YANG L, YANG Q H, CAO Y J, WEN C Y, ZHENG J K. Correlations between specific combining ability, heterosis and genetic distance in hybrid rice. Genetics, 2009, 31(8): 849-854. (in Chinese)
[45] Jiang Y, Schmidt R H, Zhao Y, Reif J C. A quantitative genetic framework highlights the role of epistatic effects for grain- yield heterosis in bread wheat. Nature Genetics, 2017, 49(12): 1741-1746.
[46] Romero N J A, Willcox M, Burgueño J, Romay C, Swarts K, Trachsel S, Preciado E, Terron A, Delgado H V, Vidal V, Ortega A, Banda A E, Montiel N O G, Ortiz-Monasterio I, Vicente F S, Espinoza A G, Atlin G, Wenzl P, Hearne S, Buckler E S. A study of allelic diversity underlying flowering-time adaptation in maize landraces. Nature Genetics, 2017, 49(3): 476-480.
[47] Eshed Y, Baum S F, Perea J V, Bowman J L. Establishment of polarity in lateral organs of plants. Current Biology, 2001, 11(16): 1251-1260.
[48] Eshed Y, Izhaki A, Baum S F, Floyd S K, Bowman J L. Asymmetric leaf development and blade expansion inare mediated by KANADI and YABBY activities. Development, 2004, 131(12): 2997-3006.
[49] Hoopes G M, Hamilton J P, Wood J C, Esteban E, Pasha A, Vaillancourt B, Provart N J, Buell C R. An updated gene atlas for maize reveals organ-specific and stress-induced genes. The Plant Journal, 2019, 97(6): 1154-1167.
[50] Stelpflug S C, Sekhon R S, Vaillancourt B, Hirsch C N, Buell C R, De Leon N, Kaeppler S M. An expanded maize gene expression atlas based on RNA sequencing and its use to explore root development. Plant Genome, 2016, 9(1): 38-54.
[51] Wu X, Li Y, Shi Y, Song Y, Zhang D, Li C, Buckler E S, Li Y, Zhang Z, Wang T. Joint-linkage mapping and GWAS reveal extensive genetic loci that regulate male inflorescence size in maize. Plant Biotechnology Journal, 2016, 14(7): 1551-1562.
[52] Peiffer J A, Romay M C, Gore M A, Flint-Garcia S A, Zhang Z, Millard M J, Gardner C A, Mcmullen M D, Holland J B, Bradbury P J, Buckler E S. The genetic architecture of maize height. Genetics, 2014, 196(4): 1337-1356.
[53] Peiffer J A, Flint-Garcia S A, De Leon N, Mcmullen M D, Kaeppler S M, Buckler E S. The genetic architecture of maize stalk strength. PLoS One, 2013, 8(6): e67066.
[54] Wang Q J, Yuan Y, Liao Z, Jiang Y, Wang Q, Zhang L, Gao S, Wu F, Li M, Xie W, Liu T, Xu J, Liu Y, Feng X, Lu Y. Genome-wide association study of 13 traits in maize seedlings under low phosphorus stress. Plant Genome, 2019, 12(3): 1-13.
[55] Dong M Y, Lei L, Fan X W, Li Y Z. Analyses of open-access multi-omics data sets reveal genetic and expression characteristics of maize ZmCCT family genes. AoB Plants, 2021, 13(5): plab048.
[56] Zhu X M, Shao X Y, Pei Y H, Guo X M, Li J, Song X Y, Zhao M A. Genetic diversity and genome-wide association study of major ear quantitative traits using high-density SNPs in maize. Front Plant Science, 2018, 9: 966.
Genome-Wide Association Analysis of Yield and Combining Ability Based on Maize Hybrid Population
LI ZhouShuai, DONG Yuan, LI Ting, FENG ZhiQian, DUAN YingXin, YANG MingXian, XU ShuTu, ZHANG XingHua, XUE JiQuan
College of Agronomy, Northwest A&F University/Key Laboratory of Biology and Genetic Improvement of Maize in Arid Area of Northwest Region, Yangling 712100, Shaanxi
【】By analyzing the yield of the hybrids from the inbred lines bred from the Shaan A and Shaan B group, the combining ability of the inbred lines were evaluated, genome-wide association analysis, and mining associated loci for yield and its combining ability conducted. It will provide references for improving maize inbred lines selected from Shaan A group and Shaan B group and applying them in varieties breeding. 【】Based on NCⅡ genetic design, 85 excellent inbred lines from Shaan A group and Shaan B group were used to construct a hybrid population containing 246 F1. Then, the yield of the hybrid population was tested in three environments to evaluate their general combining ability (GCA) and special combining ability (SCA). Using the 6H90K maize array to detect the parental genotypes, 63 879 high-quality SNPs were obtained, which were used to analyze the genetic characteristics of parental lines. According to the parental genotypes, 55 951 high-quality SNPs were inferred in the hybrid population for genome-wide association analysis of hybrid yield, GCA, and SCA using additive model and non-additive model. Meanwhile, candidate genes around the significant SNPs were screened and annotated based on the maize B73 reference genome.【】The yield in the three environments accorded to the normal distribution with wide variation, the broad-sense heritability of yield was 59.04%, and the environmental effect was significant. There was significant positive correlation between hybrid yield and combining ability, and the correlation between hybrid yield and SCA (=0.95) was higher than that between hybrid yield and GCA (=0.62). The genetic characteristic of Shaan A group and Shaan B group was different, and inbred lines from Shaan A group have higher general combining ability. Totally, five, seven and nine significant SNPs were detected (-log10()>3.86) for GCA, hybrid yield and SCA, respectively. Among them, four SNPs were co-located in hybrid yield and SCA. Ultimately, 17 associated SNPs were anchored. Dominant allele analysis of different trait-associated loci showed that four GCA-associated SNPs were controlled by additive effects, and the F1BLUE-associated loci could be divided into 4 types mainly by the dominant effect, and the heterozygous genotype is the favorite allele or sub-optimal allele for yield in F1. Through functional annotation, the candidate genes were specifically expressed in maize growth and kernel establishment, for example,andwere related to maize kernel development. 【】Based on this study, we consider that GCA and SCA jointly affect the yield of hybrids, and the effect of SCA is greater. Moreover, GCA and SCA may have different genetic basis, and GCA can be increased with the accumulation of favorable alleles. Using the genome-wide association analysis in the F1hybrid population can carry out genetic analysis related to combining ability, mine the genetic loci related to yield and combining ability, and accelerated the application of the associated loci in molecular breeding.
maize; hybrids; general combining ability; special combining ability; genome-wide association analysis

10.3864/j.issn.0578-1752.2022.09.001
2021-12-10;
2022-02-08
国家现代农业产业技术体系建设专项(CARS-02-77)
李周帅,E-mail:zhoushuai.li@foxmail.com。通信作者薛吉全,E-mail:xjq2934@163.com。通信作者张兴华,E-mail:zhxh4569@163.com
(责任编辑 李莉)
猜你喜欢
作物学报(2022年12期)2022-10-14
西南农业学报(2022年7期)2022-09-30
中国种业(2022年8期)2022-08-19
江苏农业科学(2022年6期)2022-04-15
安徽农学通报(2022年6期)2022-04-07
新课程·下旬(2018年8期)2018-11-10
科学导报(2018年47期)2018-05-14
农家科技下旬刊(2017年6期)2017-07-05
中学生理科应试(2016年4期)2016-11-19
农民致富之友(2016年5期)2016-10-21
