
苏沃雷生关键中间体的连续流微反应合成工艺研究*
2022-05-26 14:13金艳娟张安林
广州化工 2022年9期
金艳娟,张安林,,许 慧,2,马 宁
(1 湖南华腾制药有限公司,湖南 长沙 410006;2 长沙创新药物工业技术研究院有限公司,湖南 长沙 410006;3 长沙医学院药学院,湖南 长沙 410219)
苏沃雷生是以食欲素受体作为靶点的口服食欲素受体1与食欲素受体2双活性的受体抗结剂安眠药。该药物通过阻断食欲素的这两种受体的方式来影响脑部的睡眠和觉醒系统,通过睡眠-觉醒系统所形成的自发影响来实现优化睡眠质量以及治疗失眠病症,同时缓解白天易疲劳、精力不足、神思忧郁以及有失眠症的病人可能存在的注意力不集中、学习困难和记忆衰退[1-5]。该药物已经于2014年被美国FDA批准用来改善睡眠质量和治疗成人失眠症,而日本等一些国家也对这款药物完成了注册和预注册。由于副作用相对较轻,所以该药有着十分广阔的应用发展前景[6-10]。 但是,该药物的实际制备工艺中存在很多挑战,它们的合成路线繁琐,总体收率较低,多个核心中间体的分离纯化需要采用柱色谱或柱层析,而且生产制备工艺过程往往涉及到危险剧毒化学品和高危化学反应,同时还产生很多废水以及副产物,非常容易造成环境污染。这些因素极大程度上制约了其大规模工业化生产和应用[11-14]。苏沃雷生核心中间体5-氯-2-[(5R)-六氢-5-甲基-1H-1,4-二氮杂卓-1-基]苯并恶唑的化学结构式如图 1 所示。

图1 5-氯-2-[(5R)-六氢-5-甲基-1H-1,4-二氮杂卓-1-基]苯并恶唑的结构
该中间体含有一个手性碳中心,根据构造手性碳中心的途径的不同,目前已经见诸报道的制备工艺主要分为两类:手性源以及手性拆分合成法[15-18]。

图2 路线一Fig.2 Synthetic route 1
如图2所示,Mangion等研究人员[19]采取生物催化的氨基转移技术获得了对映选择性过量(ee值)大于98%的R-5-氯-2-(5-甲基-[1,4]二氮杂环庚-1-基)苯并恶唑,该步反应的收率也可以超过70%。在提升收率的大前提下,相较于过渡态重金属催化型路线而言,这条工艺路线的总体收率为43%,属于环境友好型工艺。但是,当前生物催化剂的成本价格极其昂贵,同时还很难买到和量产,这严重阻碍了该工艺线路在工业规模化生产的应用前景。

图3 路线二Fig.3 Synthetic route 2
而图3所示的是Carl A Baxter等[20]于2011年公开的一个大量制备苏沃雷生原料药的生产工艺。该工艺中所采用的合成方法绕开了脱保护基的反应过程,杂质的生成量明显减少了,从而规避了采取色谱柱法提纯的步骤,减少了氨基脱CBZ保护基的反应步骤以及最终的偶联反应,从而使得制备工艺线路短而精,效率更高。优化了原始工艺路线,采用传统的手性拆分分离途径,使用手性拆分剂从外消旋化合物5-氯-2-(5-甲基-[1,4]-二氮杂环庚-1-基)苯并恶唑中获得R-5-氯-2-(5-甲基-[1,4]-二氮杂环庚-1-基)苯并恶唑,其与5-甲基-2-[1,2,3]噻唑-2-基-苯甲酸反应获得最终产物苏沃雷生原料药。相对图2所示方案而言,此解决方案将为制备手性化合物R-5-氯-2-(5-甲基-[l,4]-氮杂环庚-1-基)苯并恶唑的工业化规模生产提供了更大的可能性,也是第一次报道实现了千克级别生产的合成工艺路线。该路线用到硫光气、光气以及甲基乙烯基酮,这些都属于剧毒品,对环境和生产设备带来极大考验和污染,工业化生产也存在诸多安全风险。
在过去的数十年时间里,许多实践案例证明了微反应器的效率远远超越批处理工艺以及现有的大体量型连续反应器[21-23]。这些微反应设备主要提供条件可控以及高通量的制备方法且存在下列诸多优势:①装置占用空间小,物料能够迅速混合以实现高效的传质传热以及生产工艺参数的精确控制,几乎不存在放大效应;过程本质安全,能够实现连续不间断操作,不仅时空效率高,而且还能节约劳动力成本。在化药合成领域有近20%的生产工艺能够通过连续流微通道反应技术实现在工艺安全性以及原子经济性等诸多方面的提高和优化[24-29]。在美国,微通道反应技术研究应用领域已经从微加工逐渐拓展成为一门有独立概念的新学科。与常规的釜式反应相比较,在微通道反应器中完成的合成过程总能够在更加短的时间范围内获得纯度更高的产品。微反应器的逐渐推广应用使得化学物质的高效制备、细胞以及蛋白质的高通量筛选和反应动力学等的研究均获得了明显提升。所以,微反应器得到了广大科研工作者以及生物医药行业巨头的关注[30-31]。所以,本研究开发了一套连续流微反应器(构造如图4所示),同时将连续流微反应技术应用于恶唑衍生物 5-氯-2-[(5R)-六氢-5-甲基-1H-1,4-二氮杂卓-1-基]苯并恶唑的制备工艺中,以解决过程中的副反应和极易造成环境污染等问题。

图4 连续流微通道反应器成套装置Fig.4 Continuous flow microchannel reactor

图5 改进后的路线Fig.5 Improved route
本文在现有路线二的基础上对化合物5-氯-2-[(5R)-六氢-5-甲基-1H-1,4-二氮杂卓-1-基]苯并恶唑的合成工艺进行优化改进。以2-氨基-4-氯苯酚(SM1)作为起始物料,乙醇作为溶剂,与乙基黄原酸钾(SM-2)环合得到化合物SM-2。SM-2与卤代试剂在微通道反应器中反应得到化合物 SM-3,SM-3与N-Boc-乙二胺反应得到中间体SM-4。该恶唑化合物(SM-4)与甲基乙烯基酮和碱反应后得到SM-5的黄色固体。恶唑化合物(SM-5)经过甲磺酸(MSA)水溶液处理得到大量淡黄色固体SM-6。SM-6通过硼氢化钠还原得到SM-7。SM-7通过二苯甲酰-D-酒石酸手性拆分后获得高纯度的5-氯-2-[(5R)-六氢-5-甲基-1H-1,4-二氮杂卓-1-基]苯并恶唑(SM-8),合成路线如图5所示。优化后的路线操作简便,安全环保,收率相对较高,目标产物纯度高,适合工业化生产。
1 实 验
1.1 仪器与试剂
Waters E2695型高效液相色谱仪,Agilent6460质谱仪,Bruker AvanceⅡ400MHz核磁共振仪。康宁Advanced-FlowTM微反应器,SF1005A型Sanotac高压四氟泵,SUNDI-755W无锡冠亚高低温循环机。所用原料和试剂均为常规市售分析纯。
1.2 5-氯-2-[(5R)-六氢-5-甲基-1H-1,4-二氮杂卓-1-基]苯并恶唑的合成
1.2.1 SM-2 的合成
在10 L四口烧瓶中加入2-氨基-4-氯苯酚 SM-1(800 g, 5.57 mol),EtOH(5 L)和乙基黄原酸钾(1.79 kg,11.1 mol)。80 ℃机械搅拌4 h。后处理:TLC检测反应液,原料已反应完,显示有一个新化合物生成。将反应液倒入8 L的冰水里,加乙酸调节pH到7。将悬浊液过滤,滤饼用水(6 L)洗涤。得到的固体减压浓缩干燥,得到1.117 kg浅灰色固体产品。备注:反应液冰水淬灭后一定要调节pH到7以下。1H NMR (400 MHz, DMSO) δ: 7.31 (m, 2H), 7.53 (d,J=8.74 Hz, 1H);LC-MS:Calculated Mw=185.63 g/mol,found m/z[M+1]+=186.89。
1.2.2 SM-3 的合成
精确称取化合物 SM-2(1.117 kg, 6.04 mol),DMF(560 mL)和DCM(11 L)充分混合制备成均匀液体备用。微反应器设置并冷却到5 ℃以下,将配置好的SM-2溶液、二氯亚砜SOCl2(1120 mL)以及缚酸剂以设定流量缓慢泵入微通道反应器。反应液流出后直接进倒入10 L的NaHCO3冰水溶液中并静置分液。水相用DCM(3 L)萃取,合并有机相,用水洗涤一次,硫酸钠干燥,减压浓缩得到1.1 kg红棕色液体产品。1H NMR (400 MHz, CDCl3) δ: 7.67 (d,J=2.1 Hz, 1H), 7.44 (d,J=8.8 Hz, 1H), 7.34 (dd,J=8.8, 2.1 Hz, 1H);13C NMR (100 MHz, CDCl3) δ:110.9, 120.2, 126.1, 131.0, 142.0, 149.9, 152.34. LC-MS:Calculated Mw=188.01 g/mol,found m/z[M+1]+=189.23。
1.2.3 SM-4 的合成
以DCM(2 L)为溶剂, 分别配制成一定浓度的SM-3(722 g, 3.87 mol)、N-Boc-乙二胺(650 g, 4.06 mol)和三乙胺(586 g,5.81 mol)溶液。反应温度保持在5~10 ℃之间,将三种原料溶液以设定流量泵入微反应器系统中,TLC显示原料已反应完,显示有两个新化合物生成。将反应液直接注入15 L冰水中,搅拌2 min后静置分液。水相用DCM(3 L)萃取一次,合并有机相,用水(5 L)反洗一次,硫酸钠干燥,减压浓缩得到的黄色固体用EA:PE(400 mL:2000 mL)打浆(4 h)。抽滤,滤饼用石油醚(1 L)洗涤。减压浓缩滤饼得到850 g淡黄色固体产品。1H NMR (400 MHz, DMSO-d6) δ ppm 8.17 (brs, 1H), 7.37 (d,J=8.40 Hz, 1H), 7.3(s, 1H), 7.02(dd,J=8.4 Hz, 1.68 Hz, 1H), 6.94 (brs, 1H), 3.18 (q,J=6.05 Hz, 2H), 1.39(s, 9H);13C NMR (100 MHz, DMSO-d6) δ 155.7, 148.1, 118.2, 114.2, 110.2, 101.5. LC-MS:Calculated Mw=311.76 g/mol,found m/z [M+1]+=313.05。
1.2.4 SM-5 的合成
以DMF(2 L)为溶剂,分别配制成一定浓度的SM-4(558 g, 1.794 mol)、DBU(327.6 g,2.152 mol)和甲基乙烯基酮(150 g, 2.152 mol)溶液。微反应器冷却到0~5 ℃,按照设定顺序将三种原料溶液以设定流量泵入微反应器系统中。后处理:TLC显示原料基本反应完,显示有两个新化合物生成。将反应液直接注入到4 L水中,有大量的固体生成,将悬浮液搅拌10 min后(冷却到20 ℃)过滤,滤饼用水(4 L)洗涤,后75 ℃烘干得到507 g淡黄色固体产品。1H NMR (400 MHz, CDCl3) ppm: 1.42 (brs. 9H), 2.24 (s, 31), 2.98(t,J=6.72 Hz, 2H), 3.47(d,J=5.38 Hz, 2H), 3.73(t,J=5.88 Hz,2H), 3.83(t,J=6.55 Hz, 2H), 4.93(brs, 1 H), 7.02 (dd,J=8.40, 1.68 Hz, 1H), 7.19 (d,J=8.74 Hz, 1H), 7.35 (d,J=1.68 Hz, 1H);LC-MS:Calculated Mw=381.85 g/mol,found m/z [M+1]+= 383.04。
1.2.5 SM-6 的合成
在3 L三口烧瓶中加入化合物 SM-5(300 g, 0.79 mol)和THF(2.5 L)。冷却到5 ℃左右,缓慢加入甲磺酸(MSA)(151.3 g, 1.58 mol),后68 ℃搅拌6 h,TLC检测没有反应完,补加甲磺酸(75.7 g,0.79 mol)并继续搅拌12 h。后处理:TLC显示原料基本反应完,显示有一个新化合物生成。将反应液冷却过滤,滤饼用THF(200 mL)洗涤,减压浓缩除去溶剂得到371 g淡黄色固体产品。1H NMR (400 MHz, DMSO-d6) ppm: 2.16(s, 3H), 2.39(s, 6H), 2.95 (brs, 2H), 3.17(d,J=8.04 Hz, 2 H), 3.56~3.65 (m, 2H), 7.08 (d,J=7.73 Hz, 1H), 7.37(brs, 1 H),7.46(d,J=8.40 Hz, 1 H),7.87(brs, 3H);LC-MS:Calculated Mw=281.74g/mol,found m/z [M+1]+=283.15。
1.2.6 SM-7 的合成
在1 L三口烧瓶中加入化合物SM-6(80 g, 169.2 mol),EtOH(800 mL)和NaOAc(13.84 g,169.2 mmol)。冷却到10 ℃左右,搅拌10 min,后加入AcOH(117.2 mL)并搅拌20 min。后分批加入硼氢化钠(7.68 g,101.5 mmol), 10~15 ℃反应2 h。后处理:1H NMR显示原料反应完,有目标产物生成。将反应液倒入冰水中,用5M的NaOH水溶液调节pH到10~11。后DCM(1.5 L×2)萃取。合并有机相,用水(1 L)反洗一次,硫酸钠干燥,有机相减压浓缩得到40 g棕色固体产品。1H NMR (400 MHz, CDCl3) ppm: 1.13 (d,J=6.39 Hz, 3H), 1.50~1.66 (m, 1H), 1.77 (brs, 1H), 1.90~2.05 (m, 1H), 2.79 (ddd,J= 9.49, 6.30, 3.02 Hz, 1H), 2.88~3.06 (m, 1H), 3.22 (dt,J=14.03, 3.23 Hz, 1H), 3.55~3.72 (m, 2H), 3.77~3.97 (m, 2H), 6.92(dd,J=8.23, 1.51 Hz, 1H), 7.11(d,J=8.40 Hz, 1H), 7.28(d,J=1.34 Hz, 1H);LC-MS:Calculated Mw=265.74 g/mol,found m/z [M+1]+=267.02。
1.2.7 SM-8 的合成
在1 L单口烧瓶中加入手性化合物二苯甲酰-D-酒石酸(91.02 g,254.2 mmol)和THF(429.2 mL)用恒压滴液漏斗滴加化合物SM-7(37 g,139.6 mmol)的DCM(42.9 mL)和THF(185 mL)的混合液。室温搅拌14 h。后处理:将反应液过滤,用100 mL THF洗涤滤饼,干燥得到29 g白色固体盐。用甲醇(200 mL)拆分得到15 g白色固体盐(97%纯度),又用甲醇(100 mL)拆分得到9 g 白色固体盐(98%纯度)。与之前做的12.5 g白色固体盐合并,加入到200 mL二氯甲烷和80 mL水中,用5 M的氢氧化钠水溶液调pH到10。静置分液,水相用DCM(100 mL)萃取一次,合并的有机相用硫酸钠干燥,浓缩得到10.5 g白色固体产品。1H NMR (400 MHz, CDCl3) ppm: 1.19(d,J=6.39 Hz, 3H), 1.62~1.72 (m, 1H), 1.98~2.12(m, 1H), 2.85 (dd,J=9.83, 6.47, 3.19 Hz, 1H), 3.01(dd,J=13.95, 10.92, 3.36 Hz, 1H), 3.28(dt,J=14.12, 3.33 Hz, 1H), 3.61~3.77(m, 2H), 3.85~4.04(m,2H), 6.98(dd,J=8.40, 16.8 Hz, 1H), 7.17(d,J=8.40 Hz, 1 H), 7.33(d,J= .68 Hz, 1H);LC-MS:Calculated Mw=265.74 g/mol,found m/z [M+1]+=267.12。
2 结果与讨论
2.1 卤代试剂对化合物SM-3的影响

图6 SM-3的连续流合成工艺流程图Fig.6 Continuous flow synthesis process of SM-3
几乎所有卤代试剂都是对空气敏感,易挥发以及环境极不友好的化学物质。在该反应中,二氯亚砜等是挑选作为SM-2中恶唑环的巯基卤代试剂,而这一步卤代反应是整个后续工艺流程中对产物收率和纯度起决定性作用的步骤。由于该反应过程存在放热现象,而且卤代试剂与底物反应后会释放出盐酸气体,所以卤代物的种类以及反应条件的可控性等对产率和纯度都会产生较大影响。因此,这一步以三乙胺作为盐酸的缚酸剂,反应采用连续流微反应系统来完成,其工艺流程如图6所示。与传统釜式反应相比,几乎没有盐酸气泄露,反应条件温和,反应速度也较快,收率也高于传统釜式反应,具体的工艺流程如图6所示。同时考察了二氯亚砜、草酰氯、光气、三氯氧磷对化合物 SM-3 纯度和收率的影响。从表1可看出,二氯亚砜用量至1.5 eq时,SM-2可以完全转化。而其它三种卤代试剂在同样时间周期内需要2 eq才能反应完全。同时也可以发现,二氯亚砜参与的卤代反应最终收率最高,达到了94.85%。

表 1 卤代试剂种类对化合物SM-3 纯度和收率的影响
2.2 缚酸剂对化合物SM-4纯度和收率的影响
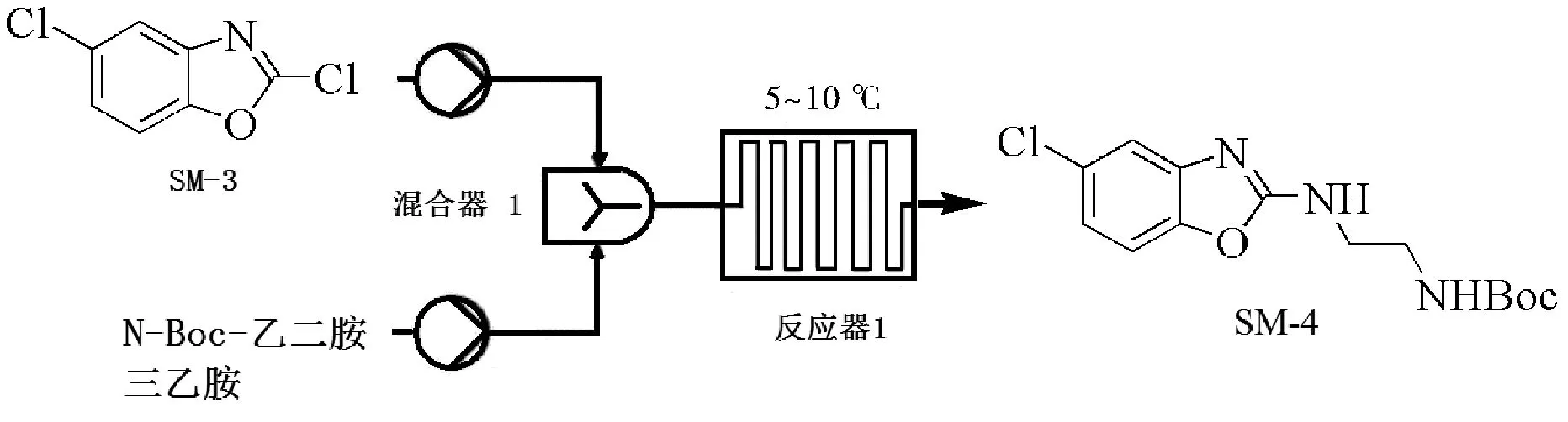
图7 SM-4的连续流合成工艺流程图Fig.7 Continuous flow synthesis process of SM-4
由于SM-3与N-Boc-乙二胺反应会生成盐酸,所以适当的缚酸剂是一方面保护原料N-Boc-乙二胺和产物SM-4中的Boc基团不被脱去,另一方面也能够催化加速SM-3与N-Boc-乙二胺的反应进程,具体的工艺流程如图7所示。考察了哌啶、N,N-二异丙基乙胺、对二甲氨基吡啶、三乙胺以及吡啶对化合物SM-4 纯度和收率的影响。

表2 缚酸剂对化合物SM-4纯度和收率的影响
从表2可以看出,缚酸剂最优的选择是三乙胺,而总体看来所列几种有机碱的效果都还不错。杂环类碱的催化收率要普遍低于普通碱,优先选择的非杂环类有机碱的优势在于它们及其盐酸盐更容易通过水洗除去,而上述几种杂环类碱及其盐酸盐相对来说不太好除尽,所以粗品从微反应器中出来后的后处理过程洗涤纯化次数会增加,这是导致收率下降的主要原因。
2.3 碱当量对化合物SM-5纯度和收率的影响

图8 SM-5的连续流合成工艺流程图Fig.8 Continuous flow synthesis process of SM-5
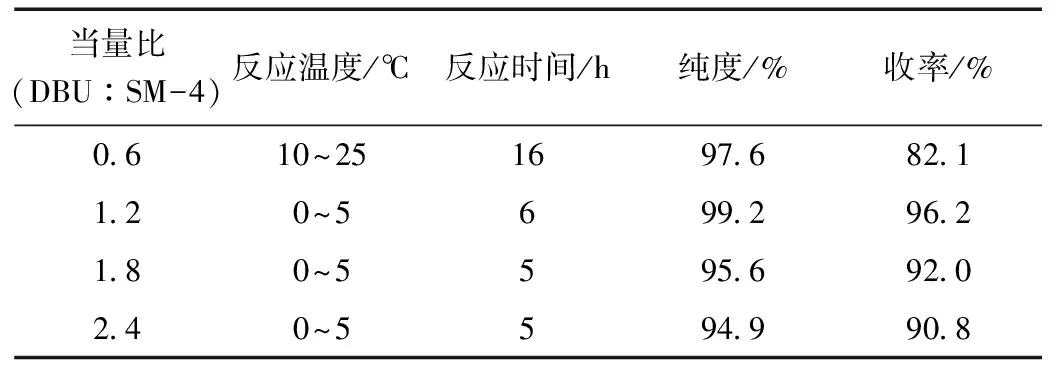
表3 碱当量对化合物SM-5收率的影响Table 3 Effect of base equivalent on the purity and yield of compound SM-5
SM-4与甲基乙烯基酮反应需要加入催化剂1,8-二氮杂二环[5.4.0]十一碳-7-烯(DBU),DBU催化剂也是反应成功的关键。该反应步骤之所以在连续流微反应器中进行主要是因为一方面:甲基乙烯基酮具有易挥发和剧毒性质,常规釜式反应器易造成污染和安全风险。另一方面该反应需要催化剂在稳定温度下才能够顺利进行。而连续流微反应器由于相对密闭的反应系统以及高效的传质传热交换性能,能够满足该步骤基于环保安全和工艺参数稳定的较高要求,具体的工艺流程如图8所示。分别以0.6、1.2、1.8、2.4当量的DBU作为合成恶唑衍生物SM-5的催化剂。从表3可看出,五种比例的DBU均可催化反应进行。0.6当量的DBU催化反应经历16 h的时间却仍然还有部分SM-4没有反应完。1.8和2.4当量的DBU虽然反应时间较短,仅需5小时,产物收率也较高,但是纯度相对1.2当量DBU的反应有明显下降。综合比较看来,1.2当量DBU催化反应温度低、时间短,产物SM-5纯度和收率均较高。
与此同时,通过对粗纯化之后的SM-5粗品核磁和质谱分析发现,如图9所示,当使用DBU量为0.6当量时,在化学位移7.02 ppm和7.37 ppm处存在SM-4的特征峰。这说明由于DBU不足会导致SM-4转化不完全,也导致后续纯化难度增加,SM-5收率降低。而通过图9发现,使用1.8 eq和2.4 eq的DBU时,由于DBU过量太多,会导致明显的杂质生成。经核磁表征并结合质谱分析,发现存在两组质荷比M/Z分别为283.2和383.1的主要杂质,推测该杂质很有可能是过量的 DBU导致反应体系碱度过高,部分SM-5中的Boc基团会被脱除直接得到了SM-6(Impurity 2),并且 Impurity 2裸露的氨基与甲基乙烯基酮继续反应得到新杂质Impurity 1。尤其是杂质Impurity 1与SM-5性质比较相近,很难通过现有的非柱层析的方法除去,这也是导致过量DBU参与的反应会引起SM-5产物收率降低的主要原因。

图9 使用不同当量的DBU反应获得的中间体 SM-5的核磁图谱和质谱图Fig.9 1HNMR and MS spectra of SM-5 intermediates obtained using different equivalent DBU
3 结 论
引入连续流微反应器并采用改进方法合成苏沃雷生核心中间体5-氯-2-[(5R)-六氢-5-甲基-1H-1,4-二氮杂卓-1-基]苯并恶唑,通过考察卤代试剂类型、缚酸剂类型、碱当量等因素对收率和纯度的影响,确定了最佳工艺条件,优化后的工艺制得的 5-氯-2-[(5R)-六氢-5-甲基-1H-1,4-二氮杂卓-1基]苯并恶唑纯度高,满足注册和市场销售的要求。该关键中间体的制备工艺方法具有原料易得、反应条件温和、操作简便、成本低廉、环境友好等优点,适合工业化生产。
猜你喜欢
世界农药(2022年10期)2022-11-10
分子催化(2022年1期)2022-11-02
化学工业与工程(2022年1期)2022-03-29
能源化工(2021年2期)2021-12-30
粉末冶金技术(2021年3期)2021-07-28
农药科学与管理(2021年2期)2021-03-16
中成药(2019年12期)2020-01-04
中学生数理化·高二版(2016年6期)2016-05-14
同位素(2014年2期)2014-04-16
火炸药学报(2014年3期)2014-03-20
