
自由基稀土配合物:研究现状、挑战与展望
2022-10-19 07:42严海涵张文雄
广西师范大学学报(自然科学版) 2022年5期
严海涵, 张文雄
(北京大学 化学与分子工程学院, 北京 100871)
稀土金属配合物不仅在金属有机化学、配位化学和有机合成化学等领域受到广泛关注[1-3],还在磁性材料和发光材料等方面有着独特的应用[4-6]。为了满足对配合物性质和功能的不同需要,人们往往通过对配体的设计和调节来合成不同的具有特定结构的稀土金属配合物。由于合成和表征手段的种种限制,早期的研究都集中在闭壳层配体的稀土配合物上。但随着研究的不断深入,人们发现,开壳层的有机自由基配体的引入会极大改变稀土金属配合物的性质,因此涌现了一系列对于自由基稀土配合物的研究。
如图1所示,从配体种类上看,常见的自由基稀土配合物主要可以分成以下3类:1)由多个同种自由基配体与稀土金属形成的均配物;2)单个自由基配体和其他闭壳层配体与同一稀土金属中心配位形成的混配物;3)自由基配体作为桥连配体连接2个稀土金属中心,与其他闭壳层配体共同形成的混配物。而其他类型的自由基稀土配合物,尤其是多个自由基配体与闭壳层配体同时与稀土中心配位的配合物则相对罕见。

图1 常见自由基稀土配合物的结构示意Fig.1 Common structures of radical rare-earth metal complexes
从自由基配体所带的电荷来看,中性自由基配体和自由基阴离子配体的稀土配合物在结构和性质上有着很大不同。中性自由基配体的稀土配合物往往具有比较特殊的磁性,但化学反应活性不高;而自由基阴离子配体的稀土配合物则常常有一些化学反应活性。因此,本文将按中性自由基配体和自由基阴离子配体的分类对自由基稀土配合物的研究现状、挑战进行总结、归纳和展望。
1 含中性自由基配体的稀土金属配合物

图2 常见中性自由基配体Fig.2 Common neutral radical ligands
有机自由基作为配体,需要在自身足够稳定的同时具有一定的配位能力。而稳定的中性有机自由基本身类型有限,且其中具有配位能力的又以氮氧自由基居多,因此绝大多数中性自由基配体的稀土金属配合物都使用氮氧自由基作为配体,只有少数情况下会使用四嗪自由基等其他自由基配体(图2)。同时,以咪唑氮氧化物为核心骨架的一系列氮氧自由基配体在自由基稀土配合物中最为常见,而较为常规的烷基或芳基氮氧化物自由基作为配体的情况则相对少见。
在中心金属的选择上,为了方便研究稀土的4f电子与自由基的相互作用,人们往往首先选用Gd作为中心金属。而在后续研究中需要单独对自由基配体进行磁学表征时,人们会选择抗磁的La和Lu以消除稀土4f电子的影响。此外,一些Tb和Dy等稀土的配合物会表现出单分子磁体性质,因此有时也会成为重点研究的对象。
1987年,Benelli等[7]首次分离并表征了稳定的自由基稀土配合物。他们使用咪唑氮氧化物自由基NITPh与二水合三(六氟乙酰丙酮)基钆[Gd(hfac)3·2H2O]在正己烷中反应,重结晶后得到配合物[Gd(NITPh)2(hfac)3](1)的晶体(图3)。由于配体的空间位阻不明显,Gd中心可以保持配位数较高的八配位、变形十二面体构型。通过对1进一步的磁性测试,他们发现自由基与Gd中心的未成对电子之间有弱的铁磁相互作用,而2个自由基之间则为反铁磁相互作用。这一结果首次揭示了稀土离子的未成对4f电子和配体上的自由基在晶体中的相互作用,为科学家们此后对自由基配体-稀土分子基磁体的大量研究打下了基础。在后续研究中,通过对咪唑氮氧化物自由基配体的改造,Benelli等[8-10]进一步合成了不同配位模式的自由基Gd配合物。例如,当使用位阻更小的咪唑氮氧化物自由基NITEt或NITiPr时,可以得到氮氧配体桥连2个Gd中心形成的一维链状结构2或3(图4)。在该结构中,Gd中心仍保持八配位变形十二面体构型,而氮氧配体的2个O原子则分别与2个Gd中心配位,桥联形成一维链状结构。
如果向咪唑氮氧化物中引入其他配位基团,配合物的结构则可以发生进一步变化[10]。例如,引入2-吡啶基可以得到O原子与吡啶N原子与Gd中心螯合配位的单核配合物4,而引入4-吡啶基则会使得吡啶N原子与另一分子中的Gd中心配位,从而得到二聚体配合物5(图5)。在这些配合物中,Gd中心都仍然保持八配位变形十二面体构型。磁性测试表明,这些配合物中Gd的4f电子与自由基之间都有铁磁耦合相互作用。
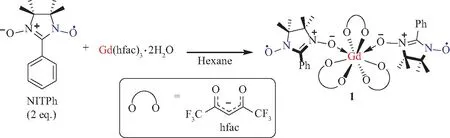
图3 配合物1的合成Fig.3 Synthesis of complex 1
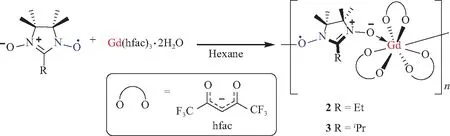
图4 一维链状自由基Gd配合物2与3的合成Fig.4 Synthesis of 1D-chain radical Gd complexes 2 and 3
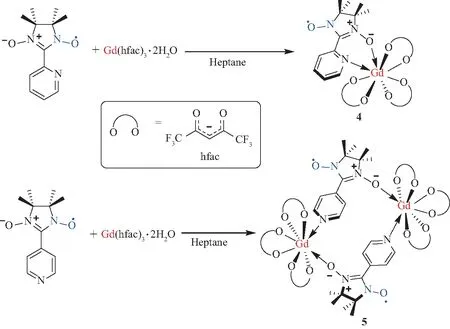
图5 吡啶基咪唑氮氧化物自由基Gd配合物4与5的合成Fig.5 Synthesis of pyridine-substituted imidazole nitroxide radical Gd complexes 4 and 5

图6 四自由基Gd配合物6的合成Fig.6 Synthesis of tetraradical Gd complex 6
为了实现更多自由基配体与稀土中心的配位,Lescop等[11-12]将稀土前体中的hfac换成更容易游离于配位层外界的高氯酸根或硝酸根,从而得到稀土中心配位多个螯合氮氧自由基配体(如NITBzImH等)的配合物。以图6为例,在产物6的阳离子部分[Gd(NITBzImH)4]3+中,Gd中心同时与4个螯合氮氧自由基配体配位。类似方法也被用来合成[Gd(NITMeBzImH)4](ClO4)3和[Gd(NITBzImH)2(NO3)3]等配合物。在磁性方面,与之前的自由基Gd配合物不同,这些多个螯合氮氧自由基配体配位的Gd配合物中,自由基与Gd的4f电子间的相互作用都是反铁磁的。另外,La、Eu等其他稀土也可以用相同方法合成类似配合物。
此后,其他科学家也合成了一系列咪唑氮氧化物自由基配体的稀土配合物[1,3,13-30],由于这些配合物的结构都比较相近,在此不再赘述。值得一提的是,一些Tb和Dy的配合物具有单分子磁体性质[31]。这些单分子磁体的有效能垒虽然都比较低(一般低于50 K),但这些结果为后续对稀土单分子磁体的研究提供了一条可行的思路,即通过自由基与稀土4f电子的自旋耦合来提高单分子磁体的性能。
除了咪唑氮氧化物自由基之外,也有少数研究采用其他类型的氮氧自由基或四嗪自由基等作为配体来合成自由基稀土配合物,例如,图7所示的3种配合物分别为单齿配位的氮氧自由基Gd配合物7[32]、螯合配位的氮氧自由基Gd配合物8[33]以及一维链状结构的四嗪自由基Gd配合物9[34]。

图7 其他中性自由基配体稀土配合物Fig.7 Other neutral radical rare-earth complexes
从以上研究工作中可以发现,在中性自由基配体的稀土金属配合物中,配体的组合几乎都离不开较稳定的自由基配体以及简单的hfac或无机酸根配体。因此,这类配合物一般比较稳定,对水氧不敏感,这对于化合物的性质测试非常有利。但另一方面,这也导致了这类配合物的化学反应性比较简单。因此,对这类配合物的研究一般都集中在磁性上。同时,由于合成前体中离不开hfac或无机酸根配体,这类配合物的组成一般比较单一,配位数和配位构型往往十分接近。这使得人们很难通过配体的调控对其结构和性质进行针对性调节。为了得到配位模式更丰富、结构更多样、化学反应活性更强的自由基稀土配合物,一些科学家将目光转向了自由基阴离子配体。
2 含自由基阴离子配体的稀土金属配合物
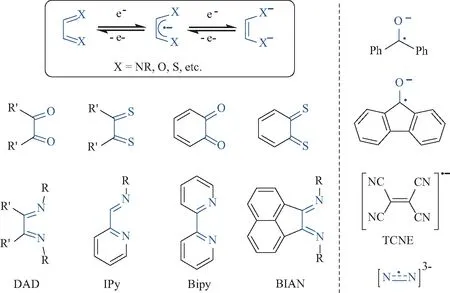
图8 自由基稀土配合物中常见的氧化还原活性配体Fig.8 Common redox-active ligands in radical rare-earth complexes
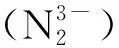
另一方面,这些常见的自由基阴离子虽然都有一定的稳定性,但它们的反应活性仍然比较强,这使得其稀土配合物具有更丰富的化学反应性。尤其是氧化还原活性配体在与Eu、Yb、Sm等具有较强单电子氧化还原能力的稀土金属配位时,常常会发生金属-配体间的单电子转移。相比于大量关于d区过渡金属与配体间电子转移过程的研究而言,4f元素的类似研究显然十分匮乏。因此,科学家们对自由基阴离子稀土配合物的研究往往更多地集中在这一单电子转移过程及相应配合物的电子结构研究上。而对于自由基阴离子与氧化还原惰性的稀土金属形成的配合物,人们也希望通过对稀土金属和其他配体的调节来调控自由基阴离子本身的反应性。另外,相比于上一章中的氮氧自由基配体,自由基阴离子配体的种类更为丰富、可调节性更强,并且其稀土配合物的合成可以脱离hfac或无机酸根配位的稀土前体,因此许多稀土单分子磁体的研究者也选择自由基阴离子配体。下面将从不同角度总结对自由基阴离子稀土配合物的研究。
2.1 自由基阴离子稀土配合物的金属-配体间单电子转移研究
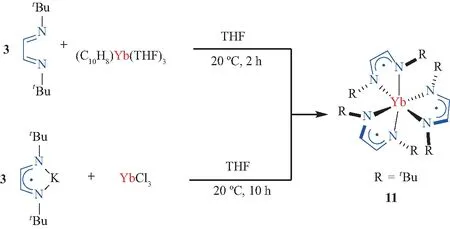
图10 均配型DAD自由基阴离子Yb配合物11的合成Fig.10 Synthesis of homoleptic radical-anionic DAD-Yb complex 11
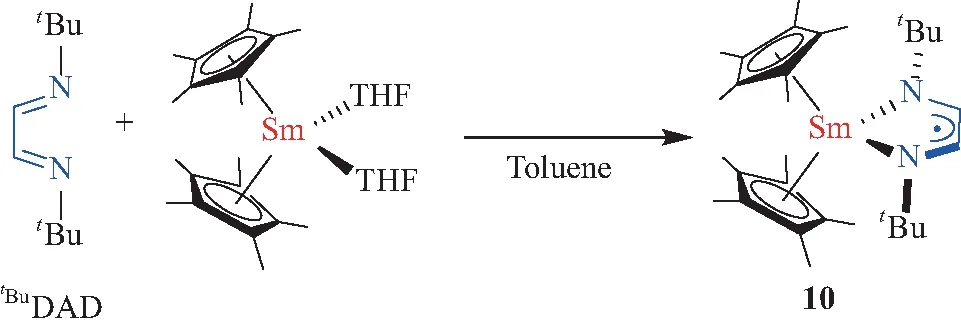
图9 DAD自由基阴离子Sm配合物10的合成 Fig.9 Synthesis of radical-anionic DAD-Sm complex 10
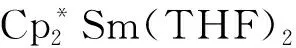
直到1995年,Cloke与Bochkarev、Trifonov、Andersen和Schumann等[39]合作,得到均配型DAD稀土配合物[Yb(tBuDAD)3](11)的晶体结构和磁学表征数据。他们发现,除了气相反应之外,通过中性DAD与萘基镱的氧化还原反应,或DAD自由基阴离子钾盐与三氯化镱的复分解反应,都可以合成配合物11,如图10。11在139 K下的晶体结构表明,Yb中心的配位构型为六配位的变形八面体,而3个DAD配体几乎完全相同,其键长也都符合DAD自由基阴离子的键长标准。因此该晶体结构对应[Yb(Ⅲ)-(DAD·-)3]的电子结构。而变温磁化率测试则表明,配合物11在不同温度下存在2种电子结构之间的转变。低温(5~15 K)下,[Yb(Ⅱ)-(DAD·-)2(DAD0)]的电子结构更加稳定,而80 K以上时,11的电子结构会完全转变为[Yb(Ⅲ)-(DAD·-)3],且自由基之间几乎没有相互作用。
此后,包括上述几位科学家在内的许多人都继续对DAD自由基阴离子的稀土配合物进行了研究。这些研究主要集中在环戊二烯基及其类似物(如茚基、芴基、硼杂苯等)的二价稀土(Eu、Yb、Sm)配合物与DAD配体间的电子转移上[40]。如图11所示,该电子转移过程可以包括DAD配体的配位和配合物的分子内氧化还原2个过程。因此,DAD配体与稀土中心之间的氧化还原电势匹配程度以及DAD配体本身的配位能力都可能对该过程产生影响。针对这2点,科学家们对该电子转移过程进行了详细研究。

图11 DAD配体与二价稀土配合物间的电子转移Fig.11 Electron transfer between DAD ligands and divalent rare-earth complexes
对于氧化电势绝对值最小的Eu(Ⅱ)而言,Moore等[41]发现,只有当DAD配体足够缺电子(如R1=C6F5, R2=Me)时,分子内氧化还原才可能在室温下发生,得到电子结构为[Eu(Ⅲ)-DAD·-]的产物;而常见的富电子DAD配体由于还原电势绝对值较高,无法接受Eu(Ⅱ)的单电子还原,只能得到电子结构为[Eu(Ⅱ)-DAD0]的产物。而对于氧化电势绝对值较大的Yb(Ⅱ),该分子内氧化还原过程显然更容易发生。Walter等[42]通过对一系列不同取代基的Yb-DAD配合物的磁性测试,证明高温下该类配合物的电子结构均为[Yb(Ⅲ)-DAD·-],且300 K下这些配合物的有效磁矩和其DAD配体的还原电势有很强的相关性;但不同配合物发生分子内氧化还原的温度会随DAD配体的取代基变化产生很明显的改变。氧化电势绝对值更大的Sm(Ⅱ)也同样满足类似的规律,例如,Cui等[43]发现,硼杂苯配体的Yb(Ⅱ)配合物与DAD反应虽然可以在室温下得到电子结构为[Yb(Ⅲ)-DAD·-]的配合物,但该反应在THF中是可逆的;而相同配体的Sm(Ⅱ)则会不可逆地得到电子结构为[Sm(Ⅲ)-DAD·-]的产物,证明Sm(Ⅱ)比Yb(Ⅱ)有更强的向DAD配体进行电子转移的趋势。
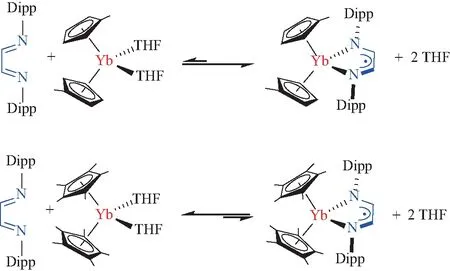
图12 大位阻DAD配体与Yb(Ⅱ)配合物间的可逆电子转移Fig.12 Reversible electron transfer between steric hindered DAD ligands and Yb(Ⅱ) complexes
另一方面,上述Yb(Ⅱ)配合物在THF中的可逆反应也表明,配位溶剂与DAD配体的竞争会影响该电子转移反应的平衡方向。这说明DAD配体的配位能力对该电子转移反应的影响也很重要。在上述Cui等[43]的工作中也提到,配位能力较弱的大位阻DAD配体无法与硼杂苯-Yb(Ⅱ)配合物发生电子转移反应。Trifonov等[44-45]则发现,DAD配体与Yb(Ⅱ)间的电子转移反应对Yb中心附近的空间位阻非常敏感。如图12所示,当Yb中心附近位阻较小时,DAD配体倾向于与Yb配位并发生单电子转移;而当Yb中心附近位阻较大时,平衡的方向则完全相反。这种空间敏感性说明DAD配体的配位能力往往不够强,很容易在与配位溶剂或其他辅助配体的配位竞争中处于不利地位,这在一定程度上限制了DAD配体的自由基稀土配合物的进一步研究。
为了进一步实现更丰富的配体组合和结构调控,科学家们也开发了一系列DAD配体的类似物,例如刚性更强、配位空间更大的二亚胺基苊(BIAN)、只有一个配位氮原子上连有取代基的亚胺基吡啶(IPy)以及2个配位氮原子上均没有取代基的联吡啶(Bipy)等。这些配体都同样具有氧化还原活性,可以生成较稳定的自由基阴离子,并且往往具有比DAD配体更强的配位能力。因此,这些配体的自由基稀土配合物不仅可以兼容除环戊二烯基以外的更多辅助配体,还可能表现出与上述DAD自由基稀土配合物不同的电子结构与反应活性。
Fedushkin等[46-48]对BIAN配体稀土配合物的单电子转移反应进行了详尽研究。如图13所示,他们发现Br配位的BIAN-Yb配合物在溶液和晶体中都是二聚体,其在高温(95 ℃)下倾向于形成[Yb(Ⅱ)-BIAN·-]2(12a)的电子结构,而低温(5 ℃)下单电子更倾向于转移到BIAN骨架上,形成[Yb(Ⅲ)-BIAN2-]2(12b)的电子结构。当配位原子由Br换成Cl时,配合物的性质会发生微妙变化。一方面,电子结构转变的温度下降到了150 K附近;另一方面,在150 K以上,二聚体中只会有一个Yb-BIAN单元发生单电子转移,从而形成[Yb(Ⅱ)-BIAN·-][Yb(Ⅲ)-BIAN2-]的电子结构。更为奇特的是,该配合物13有3种结晶方式,但只有其中一种晶型的晶体会在150 K附近发生电子结构的转变;而其他2种晶体无论温度如何,均保持[Yb(Ⅲ)-BIAN2-]2的电子结构。这些结果说明,配位环境与晶体堆积方式的微调都可能会对自由基稀土配合物的单电子转移产生很大影响。
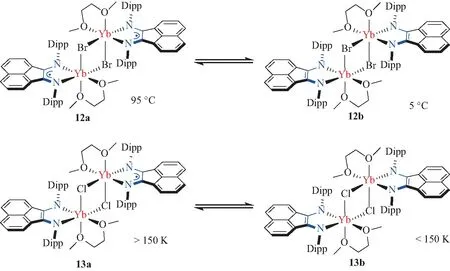
图13 不同BIAN-Yb配合物的分子内电子转移Fig.13 Intramolecular electron transfer of different BIAN-Yb complexes
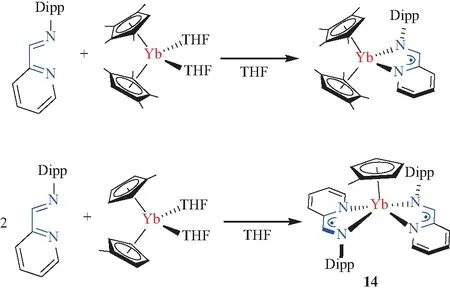
图14 IPy配体与Yb(Ⅱ)配合物间的电子转移Fig.14 Electron transfer between IPy ligand and Yb(Ⅱ) complexes
Trifonov等[49-50]则研究了IPy配体的自由基稀土配合物。他们发现,由于IPy配体的配位能力强于DAD配体,IPy稀土配合物会表现出比类似的DAD配合物强很多的电子转移倾向。如图14所示,即使Yb中心附近的位阻较大,IPy也可以与Yb中心发生不可逆的配位和电子转移反应。当Yb中心附近的位阻较小时,IPy自由基阴离子还可能发生对Cp配体的取代,生成双自由基Yb配合物14。
针对与DAD-Yb配合物类似的Bipy-Yb配合物,Schultz等[51]和Booth等[52-53]也开展了研究。他们通过变温磁化率以及X射线近边吸收(XANES)等测试手段,说明了这类配合物即使在基态下,[Yb(Ⅲ)-BIAN·-]和[Yb(Ⅱ)-DAD0] 2种电子结构的混合也非常严重,因此最好将该类配合物中的Yb中心描述为介于Yb(Ⅱ)与Yb(Ⅲ)之间的中间氧化态。此外,通过调节Bipy配体的取代基也可以显著调节两种电子结构的混合方式。
除了上述包含1, 4-二氮杂丁二烯类配体的自由基稀土配合物之外,其他的杂丁二烯类配体,例如α-二酮[54-56]、邻苯醌[29]等配体,乃至联磷杂苯配体[17]的自由基稀土配合物也有类似的电子转移反应报道。但这些配体的可修饰位点较少,导致配合物的结构拓展性不强,因此相关研究的数量相对较少,尚缺乏足够的系统性,在此不再赘述。
2.2 自由基阴离子稀土配合物的其他化学反应研究
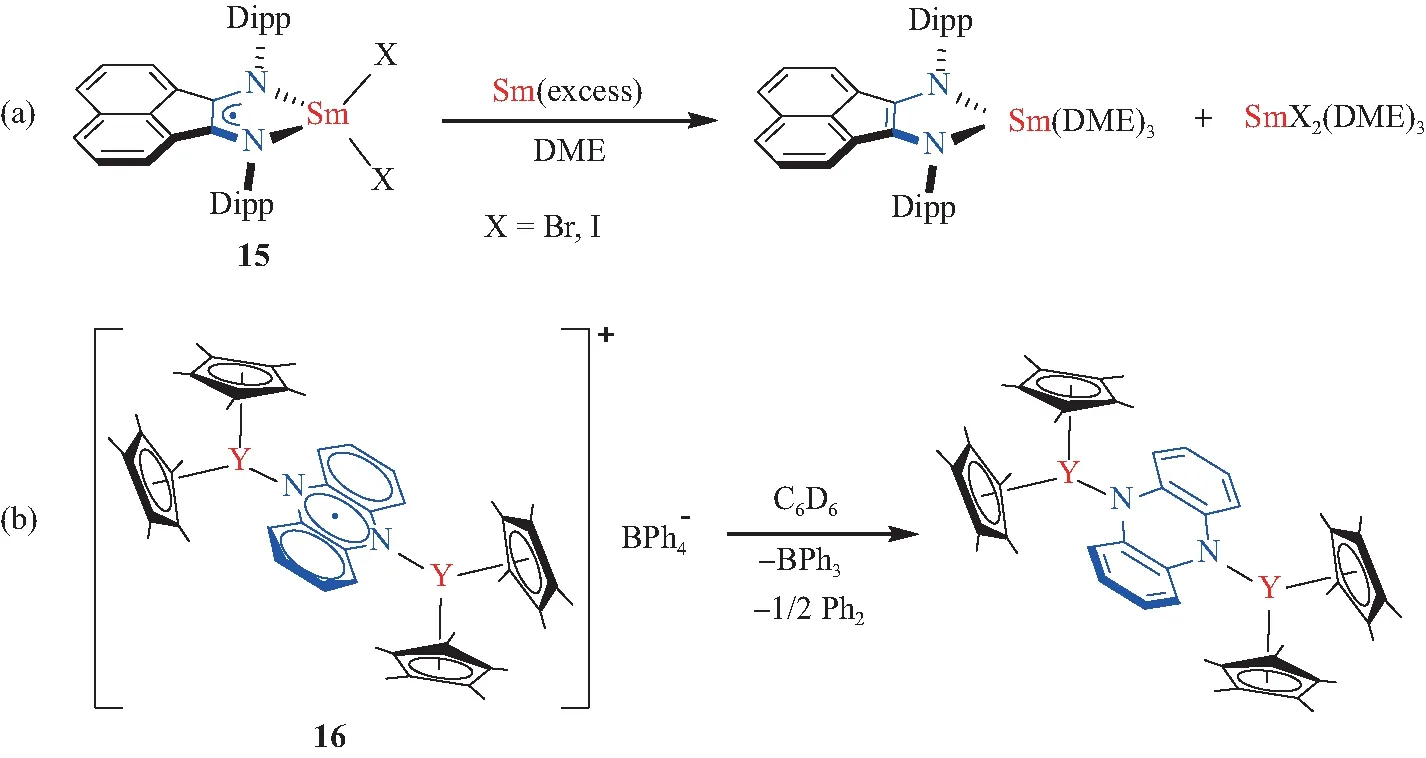
图15 自由基阴离子稀土配合物的还原反应Fig.15 Reduction of rare-earth complexes with radical anions
自由基阴离子配合物的氧化反应也有报道。例如,Ortu等[59]发现,Bipy-Ce配合物可以被二苯甲酮氧化,得到偶联的二苯甲酮桥连的双核Ce配合物17(图16)。

图16 自由基阴离子稀土配合物的氧化反应Fig.16 Oxidation of rare-earth complexes with radical anions
常见的一些配体交换反应也可以发生在自由基阴离子稀土配合物中。大位阻的DAD自由基阴离子配体由于配位能力较弱,会在配位竞争中处于不利的地位,因此配体交换反应常常导致其解离。例如,Kissel等[60]报导了DAD-Y配合物18与不同烷基锂的配体交换反应,发现即使是简单的盐复分解反应也会伴随DAD配体的解离,得不到DAD自由基阴离子配位的稀土烷基配合物(图17)。

图17 自由基阴离子稀土配合物的配体解离Fig.17 Ligand dissociation of rare-earth complexes with radical anions
只有当自由基阴离子配体的配位能力更强时,配体交换反应才会选择性地发生在其他配体上。对于DAD或类似自由基阴离子的稀土配合物来说,这种情况较少见。Fedushkin等[57]发现,BIAN-Sm配合物19可以与当量的叔丁醇钾发生盐复分解反应,没有导致BIAN配体的解离(图18),产物20为罕见的含1,4-杂丁二烯类自由基阴离子配体的稀土烷氧基配合物。
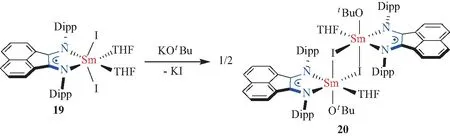
图18 自由基阴离子稀土配合物的盐复分解反应Fig.18 Salt metathesis of rare-earth complexes with radical anions

图19 芴酮自由基阴离子Sm配合物的自由基偶联反应Fig.19 Radical coupling of fluorenone radical anion-Sm complexes
此外,在自由基阴离子稀土配合物中,自由基的偶联反应也有可能发生。例如,Hou等[61-64]发现,芴酮自由基阴离子的Sm配合物可以发生自由基偶联反应,并且通过对配位溶剂的调节可以控制反应平衡的方向(图19)。当配位能力较弱的乙醚与Sm中心配位时,反应会向自由基的偶联方向进行;而当配位能力更强的THF或HMPA与Sm中心配位时,芴基自由基阴离子则更加稳定。并且,由于HMPA的配位能力过强,难以被其他配位溶剂取代,相应的配合物21无法向自由基偶联产物22转化。该反应从侧面证实了金属对酮的还原偶联反应可以通过自由基机理发生。
2.3 自由基阴离子稀土配合物的单分子磁体性质研究
自由基与稀土中心未成对4f电子间的相互作用会影响稀土配合物的磁学性质。同时,含自由基阴离子配体或中性自由基配体的稀土配合物对稀土中心上的其他辅助配体以及配合物配位构型的兼容性完全不同。因此,许多科学家对自由基阴离子稀土配合物的磁学性质进行研究,并开发了多种含有自由基阴离子的稀土单分子磁体。图20给出了其中一些代表性的例子。

图20 一些代表性的含自由基阴离子的稀土单分子磁体Fig.20 Some representative rare-earth SMMs with radical anionic ligands

Gould等[70]进一步通过对24中联嘧啶配体的修饰,研究了单分子磁体的性能与桥连自由基阴离子配体上取代基的关系。他们发现,联嘧啶配体上的取代基吸电子能力越强,稀土离子与自由基之间的自旋耦合作用就会越强,相应的单分子磁体的有效能垒也越高,这给同类稀土单分子磁体性能的提升提供了一个有效的思路。
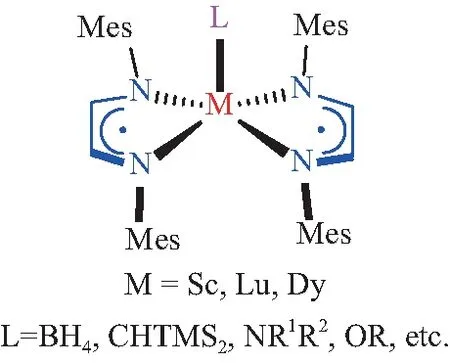
图21 高配体兼容性的双自由基稀土配合物Fig.21 Rare-earth diradical complex bearing adaptable auxiliary ligands
2.4 自由基阴离子稀土配合物的配体兼容性拓展
观察上述所有自由基稀土配合物的组成和结构,可以发现,稀土中心的选择一般没有限制,但自由基配体与闭壳层配体的组合则往往受限。一旦确定了自由基配体,配合物中可以兼容的闭壳层配体实际上寥寥无几。例如中性自由基配体往往只能兼容hfac或简单无机酸根配体,而自由基阴离子配体几乎只能与Cp类配体、卤素、配位溶剂或少数几类螯合配体进行组合。这在很大程度上限制了人们对自由基稀土配合物的组成和结构的调控,进而限制了对其化学性质和磁学性质的调节。换言之,目前自由基稀土配合物研究的瓶颈在于高配体兼容性的自由基稀土配合物的合成。
最近,Yan等[71-73]发现,通过增强配体间的π-π相互作用,可以有效抑制DAD稀土配合物的配体重分配过程,从而合成一系列稳定的双自由基稀土配合物。该体系可以兼容包括硼氢酸根、烷基、胺基、硼氧基、烷氧基、氮氧基等配体在内的大量闭壳层辅助配体,从而实现对同一系列自由基稀土配合物中辅助配体的调控(图21),并且在此基础上进一步调控了该类配合物的化学和磁学性质。
3 结语
第一个稀土元素的发现距今已有220余年,稀土金属有机化学则有近70年的历史。直到最近30年,人们才开始探索有机自由基与稀土金属的结合。因此,自由基稀土配合物这一领域仍然具有很大的发展空间。例如,对于自由基配体来说,虽然现有的自由基配体种类繁多,但如果除去中性氮氧自由基和1,4-杂丁二烯自由基阴离子,剩余的例子则寥寥无几。另一方面,对于闭壳层辅助配体来说,其与自由基配体之间的兼容性仍然是一个亟待解决的问题。只有进一步解决该问题,才能够真正实现对自由基稀土配合物的结构微调,从而更好地调控配合物的化学、磁学、光学等各方面性质。除此之外,许多传统的稀土金属配合物,例如单核稀土阳离子配合物、四核以上的稀土簇合物等结构中,都未曾引入过自由基配体。一些可能存在催化活性的自由基稀土配合物[74]也尚未得到足够充分的研究。这些问题都将成为该领域未来所必须面临的挑战。
致谢:特别感谢席振峰院士对稀土金属有机化学课题组(CCREM课题组)的无私奉献和大力支持!
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
装备制造技术(2020年3期)2020-12-25
当代陕西(2019年6期)2019-04-17
现代农业(2015年3期)2015-02-28
创新科技(2014年21期)2014-12-23
应用化工(2014年1期)2014-08-16
应用化工(2014年1期)2014-08-16
无机化学学报(2014年4期)2014-02-28
中国信息化·学术版(2013年5期)2013-10-09
郑州大学学报(理学版)(2013年2期)2013-03-11
