
平面十二配位MB8C4(M=Ca, Sr, Ba)分子轮团簇的理论研究
2023-10-17 23:51冯林雁胡晓波苗常青郭谨昌王迎进
高等学校化学学报 2023年10期
冯林雁, 胡晓波, 闫 苗, 苗常青, 陈 瑞, 郭谨昌, 王迎进
(1. 忻州师范学院化学系, 忻州 034000; 2. 山西大学分子科学研究所, 纳米团簇实验室, 太原 030006)
硼与碳虽然为元素周期表中的近邻元素, 但不同的价电子排布, 导致二者形成的化合物和材料具有显著差异. 硼典型的缺电子性赋予裸硼团簇独特的几何结构和新颖化学成键[1,2]. 其不同于块体材料, 也不同于小分子体系. 单质硼是以正二十面体笼状B12单元构成的三维材料, 而大量的实验和理论研究表明, 小尺寸硼团簇在广泛尺寸范围内均具有平面或准平面结构[3~13]. 这些结构的稳定性被归功于硼内核和外环之间的离域π/σ键, 致使硼基纳米团簇呈现双重芳香性、 反芳香性和冲突芳香的化学成键特征[14]. 随着尺寸的增加, 裸硼团簇呈现双环管状或笼状结构[15,16]. 其中, 以B 原子为中心的D7hB28(-B©B7)2-和D8hB(-9B©B-8)团簇作为平面多配位分子轮团簇的典型代表[5], 具有非同寻常的6π/6σ双重芳香性, 激励科学家不断去设计或寻找一些平面多配位分子轮团簇. 2000年, Schleyer等[17,18]提出了以主族元素C为中心的平面分子轮CB26-和CB-7团簇. 但由于C电负性大, 避免中心高配位位置, 致使这两结构均不是势能面上的全局极小. 随后, 一系列以过渡金属为中心的分子轮团簇被相继发现, 如M©B(-nM=Co, Ru, Ir, Fe, Nb, Ta)[19~24]. 目前, Ta©B-10和Nb©B-10团簇被认为是在二维平面体系中拥有最高配位数的硼分子轮.
在稳定平面多配位硼分子轮结构中, 中心原子和外围硼环必须在几何尺寸和电子计数方面相契合, 因此, 合理选择中心金属和外围硼环的原子数目是稳定分子轮结构的关键所在. 为了理解该类结构的稳定性, Wang 等[19~21]总结了设计此类结构的电子计数规则:n+x+q=K(其中,n为硼原子的数目,x为中心金属的形式价态;q为体系所带的电荷数;K为体系总的离域电子数目). 为了满足双重芳香性,K值一般为12或16. 该电子计数规则已成功应用于各种以过渡金属为中心的平面硼分子轮, 包括D8hCo©B8,D9hIr©B9,D10hTa©B-10等团簇. 化学成键分析表明, 中心过渡金属和外围硼环之间的强共价作用对稳定分子轮结构起关键性作用. 相比之下, 以主族金属或贵金属为中心的硼分子轮, 由于中心金属缺乏d轨道或d轨道全满, 致使高配位轮状结构稳定性较差. 如AlB-7和AlB-8团簇的最稳定结构为伞状结构, 而不是轮状结构[25].
目前, 化学家对平面多配位分子轮的认知远远超出以金属为中心的硼单环结构. 最近, Li等[26]通过第一性原理计算设计了LaC13团簇, 该团簇是一个具有十三配位的完美平面碳分子轮. 随后该课题组理论预测了碱金属掺杂的完美平面CsC18分子轮[27], 是目前平面体系中拥有最高配位数的团簇.Wang等[28,29]通过掺杂最大共价半径的La和Y元素, 发现十二配位LaB8C4和YB8C4金属硼碳环团簇, 进一步突破了对平面多配位团簇结构和成键的理解. 近期研究表明, 碱土金属具有灵活成键的特征, 能够产生一些新颖的几何结构和化学成键[30,31]. 受此启发, 我们推测将La/Y 元素换成与之近邻的Ca,Sr, Ba元素也可以稳定B8C4环, 形成以碱土金属为中心的轮状平面结构. 基于此, 本文主要围绕碱土金属掺杂的十二配位硼碳分子轮MB8C4(M=Ca, Sr, Ba)团簇展开, 从几何结构、 成键性质以及芳香性等方面探讨体系的稳定性, 研究结果进一步丰富了平面多配位配合物家族, 为合理设计更多该类化合物提供了一定的理论依据.
1 理论方法
在B3LYP/Lanl2dz 水平下, 采用Coalescence-Kick (CK)搜索[32,33]程序和手工搭建方法对MB8C4(M=Ca, Sr, Ba)团簇可能存在的异构体进行全局搜索, 每组体系初步计算不少于3000个异构体. 针对不同碱土金属掺杂的体系, 初步筛选出能量在209.2 kJ/mol 以内的低能量异构体, 并在B3LYP-D3/def2-TZVP水平下再次进行优化和频率计算, 确保结构是势能面上的极小结构. 为了证实能量的准确性, 也对前10个低能量异构体在CCSD(T)/def2-TZVP水平下进行了单点计算[34,35]. 采用NBO 6.0程序[36]进行韦伯键级和自然原子电荷计算. 化学成键分析采用正则分子轨道(Canonical molecular orbitals,CMOs)、 适应性自然密度划分(Adaptive natural density partitioning, AdNDP)[37]和电子定域化分析(Electrons local functions, ELFs)方法[38,39]. 为了验证体系的芳香性, 采用最近比较流行的各向磁感应电流密度方法(Anisotropy of the induced current density, AICD)绘制体系的感应电流密度图[40]. 采用Multiwfn 3.6软件[39]对分子轮结构进行电子密度拓扑分析(QTAIM). 所有计算都在Gaussian 09程序[41]下运行. 采用GaussView和Molekel 5.4程序[42]对CMOs和AdNDP结果进行可视化.

Fig.1 Global minimal structures and low-lying isomers of CaB8C4(A), SrB8C4(B) and BaB8C4(C)clusters at B3LYP-D3/def2-TZVP level
2 结果与讨论
2.1 结构与稳定性
2.1.1 MB8C4(M=Ca, Sr, Ba)团簇的低能量异构体 在B3LYP-D3/def2-TZVP水平下优化不同碱土金属掺杂的MB8C4(M=Ca, Sr, Ba)团簇的低能量异构体, 所有体系均考虑自旋单重态和三重态. 图1给出这些体系前10个低能量异构体在B3LYP 和CCSD(T)水平下的相对能量和电子态. 值得注意的是, 这两种水平下获得的异构体的能量顺序略有差异. 通常耦合簇CCSD(T)方法被认为是计算硼纳米团簇最精确的方法, 因此, 主要以CCSD(T)计算结果为主. 经CK 搜索、 结构优化和单点能校准, 最终确定CaB8C4团簇的全局极小结构为具有D4h对称性的完美轮状结构. 其中, Ca 原子位于高对称性B8C4环的中心. 在B3LYP水平下, 第二低能量异构体(a2)与基态结构的区别在于电子态不同, 其能量比基态结构高21.42 kJ/mol. 纵观这些低能量异构体, 前10个异构体中有5个是平面轮状结构, 间接表明平面轮状结构的稳定性.
在相同理论水平下, 用同族Sr原子替换中心十二配位Ca原子, 发现平面轮状结构仍为SrB8C4团簇的全局极小结构. 但在CCSD(T)水平下, 存在一个拉长状八配位异构体(b4)与轮状结构能量接近, 可能是共存异构体. 随着掺杂原子半径的增加, 以Ba为中心的轮状结构的稳定性逐渐下降. 在CCSD(T)水平下, 该结构的能量比拉长状八配位异构体(c4)高24.98 kJ/mol, 表明较大原子半径Ba与B8C4环在尺寸上无法完美匹配, 致使拉长状八配位结构与轮状结构在能量上存在激烈竞争. 此外, 也计算了以Mg为中心的分子轮结构, 发现Mg原子由于半径较小, 导致其偏离分子轮中心.
2.1.2 轮状CaB8C4和SrB8C4团簇的键长 图2 展示了轮状CaB8C4和SrB8C4团簇的键长. 对于CaB8C4团簇, 对称性为D4h, 具有相等的B—B或B—C键长. 根据Pyykkö推荐的共价键长[43], B—B单键和B=B双键键长的上限分别为0.170 和0.156 nm, B—C 单键和B=C 双键的键长分别为0.160 和0.145 nm.基于此, 轮状CaB8C4团簇的B—B键(0.157 nm)接近于B=B双键, B—C键(0.138 nm)落入B=C双键范围[图2(A)]. Ca—B 和Ca—C 键长分别为0.280 和0.275 nm, 大于Ca—B(0.256 nm)和Ca—C(0.246 nm)单键的上限值, 暗示中心金属Ca与外围B8C4环之间存在微弱共价作用. 轮状SrB8C4团簇与CaB8C4有相似的几何结构特征. 与CaB8C4团簇相比, 外围B—B 键距离略微拉长0.001 nm[图2(B)].由于Sr 原子半径大于Ca, 导致中心金属Sr 突出平面0.083 nm, 且中心金属Sr 与外围硼碳环的距离明显长于相应CaB8C4团簇的键长. 总之, 外围B8C4环的尺寸与Ca掺杂B8C4环相比, 扩大了0.004 nm. 随着金属原子尺寸的增加, 以Ba为中心的轮状结构的稳定性逐渐变差, 暗示尺寸匹配效应是环状结构稳定的关键因素之一.
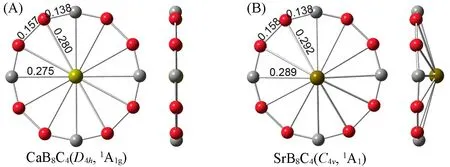
Fig.2 Optimized molecular wheel structures for CaB8C4(A) and SrB8C4(B) clusters at B3LYP-D3/def2-TZVP level
2.1.3 轮状CaB8C4和SrB8C4团簇的韦伯键级和自然原子电荷 韦伯键级数据可进一步支持上述推断.图S1(见本文支持信息)呈现了轮状十二配位CaB8C4和SrB8C4团簇的键级和自然原子电荷数据. 对于CaB8C4团簇, B—B和B—C键键级明显大于1, 分别为1.26和1.70, 暗示外围B8C4环除形成2c-2eσ键以外, 还存在离域π/σ键. B—C 键的键级明显强于B—B 键, 表明离域π/σ键的电子云主要集中在B—C键上. 相比之下, Ca—B和Ca—C有较小的韦伯键级(0.03), 相一致于其较长的键长, 暗示Ca与外围B8C4环之间离子键作用占主导. 与CaB8C4团簇相比, 轮状SrB8C4团簇除了外围B—B 键略弱于CaB8C4团簇之外, 其余键的键级均相同.
自然原子电荷分析表明, Ca/Sr原子和外围B8C4环之间存在明显的电子转移. 中心金属Ca/Sr均失去电子, 带1.80 个正电荷, 接近于Ca2+/Sr2+. 由于C 原子电负性大于B, 因此C 原子携带-1.00/-0.98|e|, 而B 原子带有0.27/0.26 |e|. 外围B8C4环共计携带-1.84 |e|, 表明中心金属和外围B8C4环之间存在较强的离子作用. 总之, 团簇MB8C4(M=Ca, Sr)可近似描述为M2+[B8C4]2-离子化合物.
2.1.4 拉长状八配位SrB8C4团簇的结构 由于拉长状八配位SrB8C4团簇与轮状结构能量相近, 且该结构为BaB8C4团簇的基态结构, 因此有必要对其结构进行简单描述. 如图3所示, 该结构是由拉长状B7C4环和两个独立的B, Sr 原子组成. 其中, B 原子位于右半环的中心, 而Sr 原子位于左半环中心上方0.209 nm处. 图3(A)和(B)分别展示了该结构的键长、 键级和自然原子电荷. 根据Pyykkö推荐的共价键长[43], 外围B—B键(0.148~0.155 nm)介于单双键之间, B—C键接近于B=C双键. 由图3(B)可知,外围B—B键级为1.21~1.58, B—C键级为1.10~1.86, 暗示外围B7C4环上除了存在2c-2eσ键之外, 还存在离域π/σ键, 且离域键电子云主要集中在B—C 单元. Sr 与外围邻近B/C 原子的键级可忽略不计(接近于0), 表明Sr—B/Sr—C键之间以离子键作用为主. 相反, 中心B与邻近B/C原子的键级为0.42~0.80, 表明B—B/B—C键之间以共价作用为主. 自然原子电荷分析表明Sr原子携带1.79个正电荷, 表明有89%的电子转移到B7C4单元, 且与Sr近邻的B/C原子带有大量电荷. 但2个价电子并未完全转移到硼碳单元, 暗示在Sr与邻近B/C原子之间存在微弱共价作用.
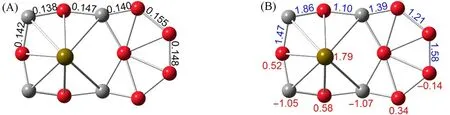
Fig.3 Calculated bond distances(in black)(A), WBIs(in blue) and natural atomic charges(in |e|, red)(B) of elongated SrB8C4(Cs, 1A′) cluster with octa-coordinated number
2.2 轮状CaB8C4团簇的成键本质
为了理解MB8C4(M=Ca, Sr, Ba)团簇的结构稳定性, 通过正则分子轨道、 AdNDP和ELF分析[37~39]来阐明这3个体系的成键本质. 鉴于轮状CaB8C4和SrB8C4团簇具有相似的成键特征, 主要以CaB8C4作为典型例子来进行化学成键分析. 团簇CaB8C4有42个价电子, 根据原子轨道类型可以将这些轨道划分成3个亚组, 如图4所示.
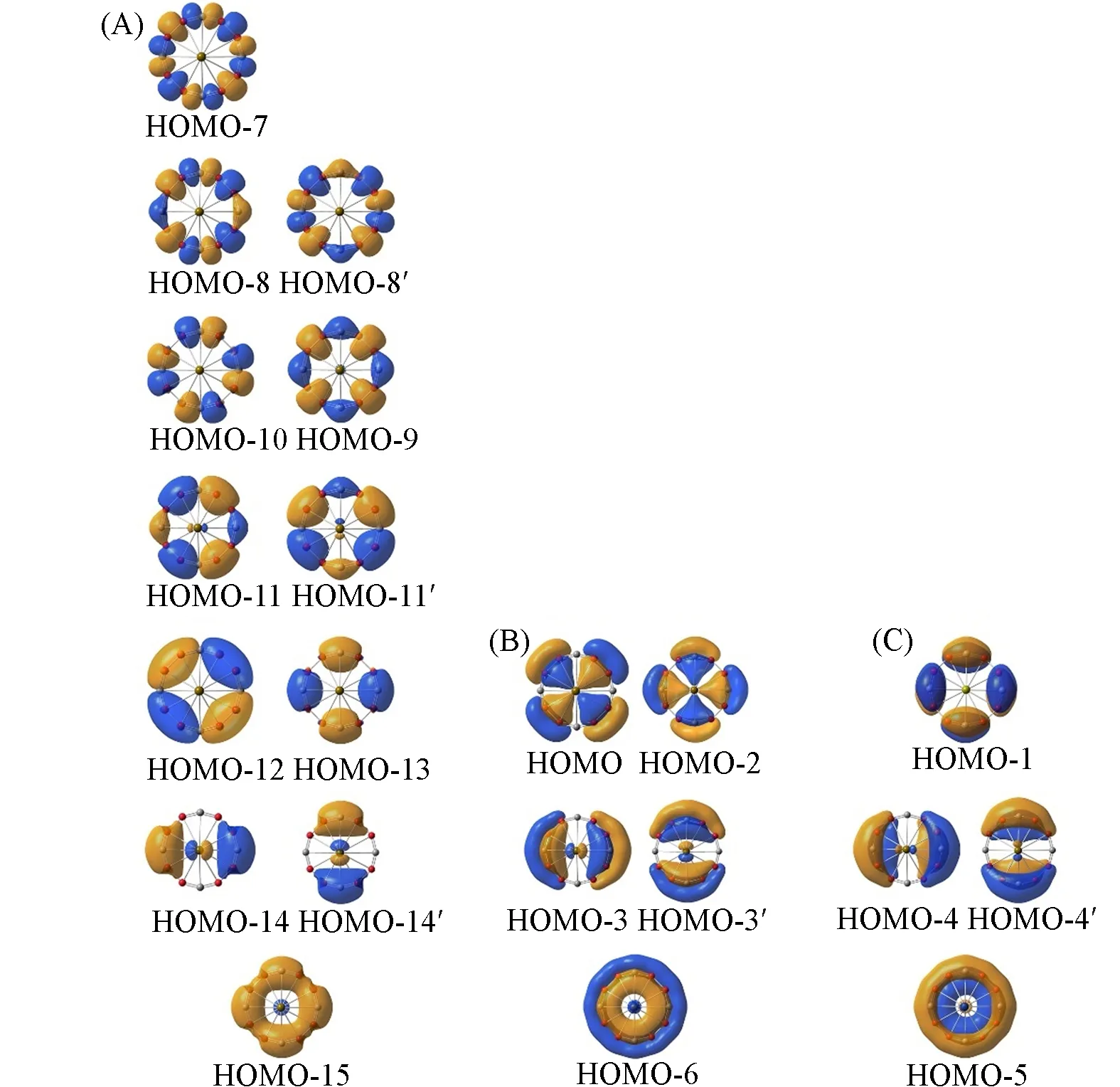
Fig.4 Occupied canonical molecular orbitals(CMOs) of dodeca-coordinated CaB8C4 cluster
图4(A)中的亚组包含12个占据轨道, 每个轨道主要基于B或C的2s/2p原子轨道形成. 这些轨道从下到上分别有0~6 个节面, 并且遵循轨道构造原则. 经轨道重组后, 这些轨道可以定域为外围B8C4环的12个2c-2e B—B或B—Cσ键. 这12个σ键共消耗体系的24个电子, 余下18个电子形成离域π/σ键.
图4(B)中的亚组包含5个离域σ轨道, 是基于B 2p轨道和Ca 3d轨道形成的. 轨道组分分析表明,HOMO 和HOMO-2 轨道中Ca 原子分别贡献3.8%(3d)和1.9%(3d), 暗示Ca 与B8C4环之间存在微弱的共价作用, 并且Ca原子表现出过渡金属性质. 这5个离域σ轨道符合休克尔(4n+2)规则, 因此, 该体系具有σ芳香性. 图4(C)中的亚组代表4个离域的π轨道, 符合休克尔4n规则, 致使该体系具有π反芳香性.
众所周知, Ca原子的价电子排布为4s2, 失去1.80个电子后s轨道上应留有少量电荷. 而NBO分析表明Ca的价电子排布发生明显变化, 为4s0.043d0.16. 在3d轨道上存在少量电荷, 且Ca 3d参与离域键的形成, 暗示Ca原子具有一定过渡金属性质. 相似的成键情形也出现在碱土金属羰基配合物中[44,45].
Boldyrev开发的AdNDP程序[35]能够直观验证复杂体系的化学成键, 目前已被广泛应用于硼基纳米团簇多中心键的研究. 为了进一步理解十二配位轮状结构的成键模式, 以CaB8C4团簇为例, 使用AdNDP程序对其进行化学成键分析, 结果如图5所示.
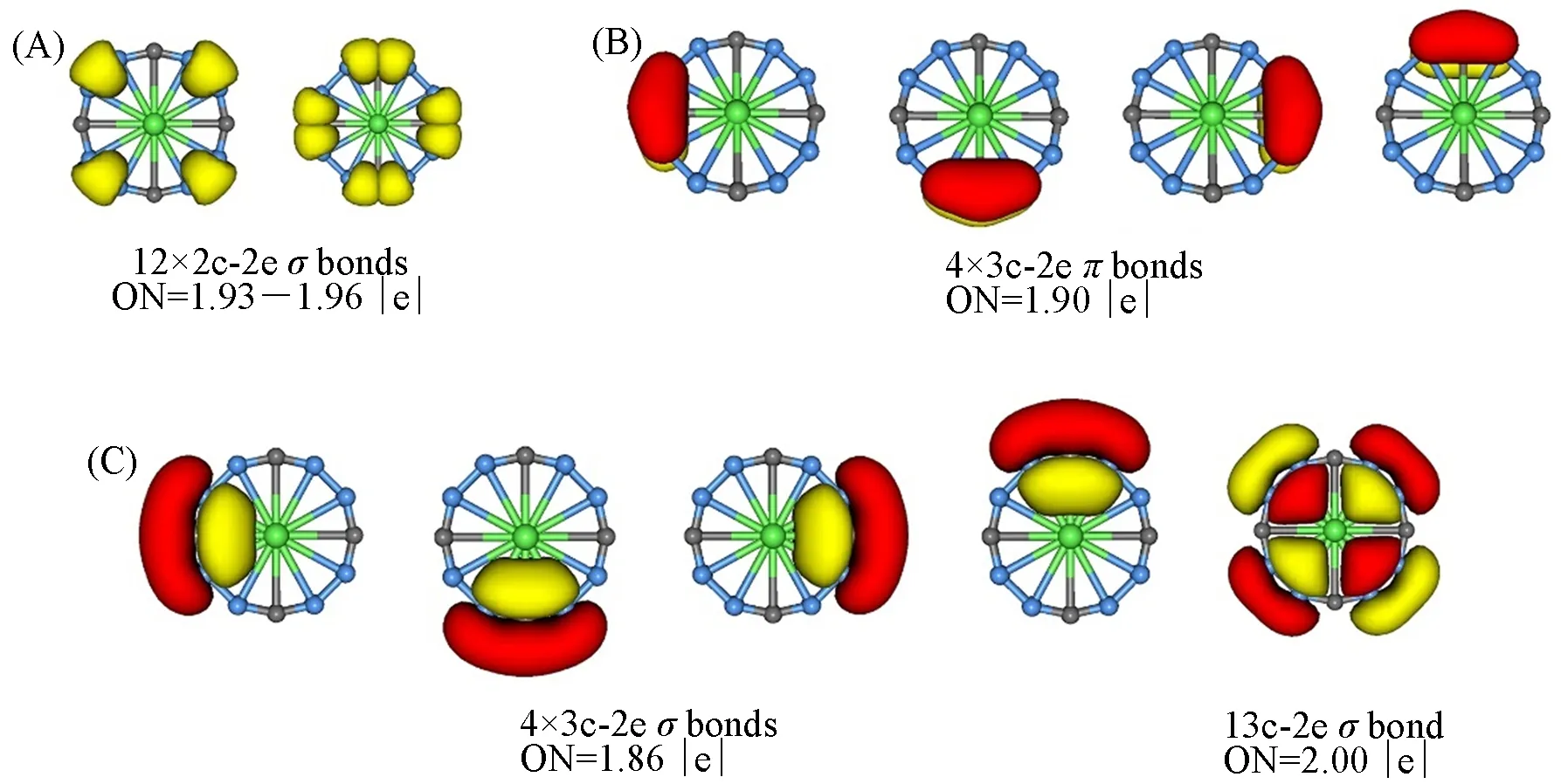
Fig.5 Bonding pattern of the CaB8C4 cluster via adaptive natural density partitioning(AdNDP) analysis
图5(A)中的亚组代表外围B8C4环上的8个2c-2e B—Cσ键和4个2c-2e B—Bσ键. 4个离域的π轨道被定域为相应的3c-2e B—C—Bπ键[图5(B)], 其占据值(ON)值为1.90 |e|, 表明中心Ca 原子与外围B8C4环之间以离子作用为主. 此外, 在B8C4环上还存在4 个离域3c-2e B—C—Bσ键和1 个离域的13c-2eσ键[图5(C)]. 其中, 3c-2eσ键的ON值明显小于3c-2eπ键, 表明Ca原子参与离域σ键的贡献大于离域π键. 总之, AdNDP成键方案很好地勾勒出轮状结构的成键图片, 证实轮状结构具有8π/10σ冲突芳香性的成键特征.
类似地, ELF数据也支持了轮状团簇的离域π/σ成键和芳香性特征(见本文支持信息图S2). 在硼基纳米团簇中, 相关文献表明双重芳香性更有利于结构呈现圆环状, 冲突芳香性则会使结构在一个方向呈现拉长状[5,14,19~24]. 然而, 目前的体系可视为是轮状结构的一个特例. 虽然它具有冲突芳香的成键特征, 但碱土金属与外围B8C4环之间的强静电作用和微弱共价作用有利于体系稳定性的提高, 打破了目前对轮状结构稳定性的认知.
为了进一步探讨轮状团簇的芳香性, 利用AICD程序[40]描绘了最近比较流行的磁感应环电流密度图. 图6(A)和(B)分别呈现了轮状CaB8C4和SrB8C4团簇高分辨率的分解σ和π环电流图. 可知, 在整个B8C4环上显示强σ环电流. 相比之下, 其π环电流明显定域在4个BCB单元上, 且电流方向与σ环电流相反, 证实了该体系冲突芳香性的成键特征.
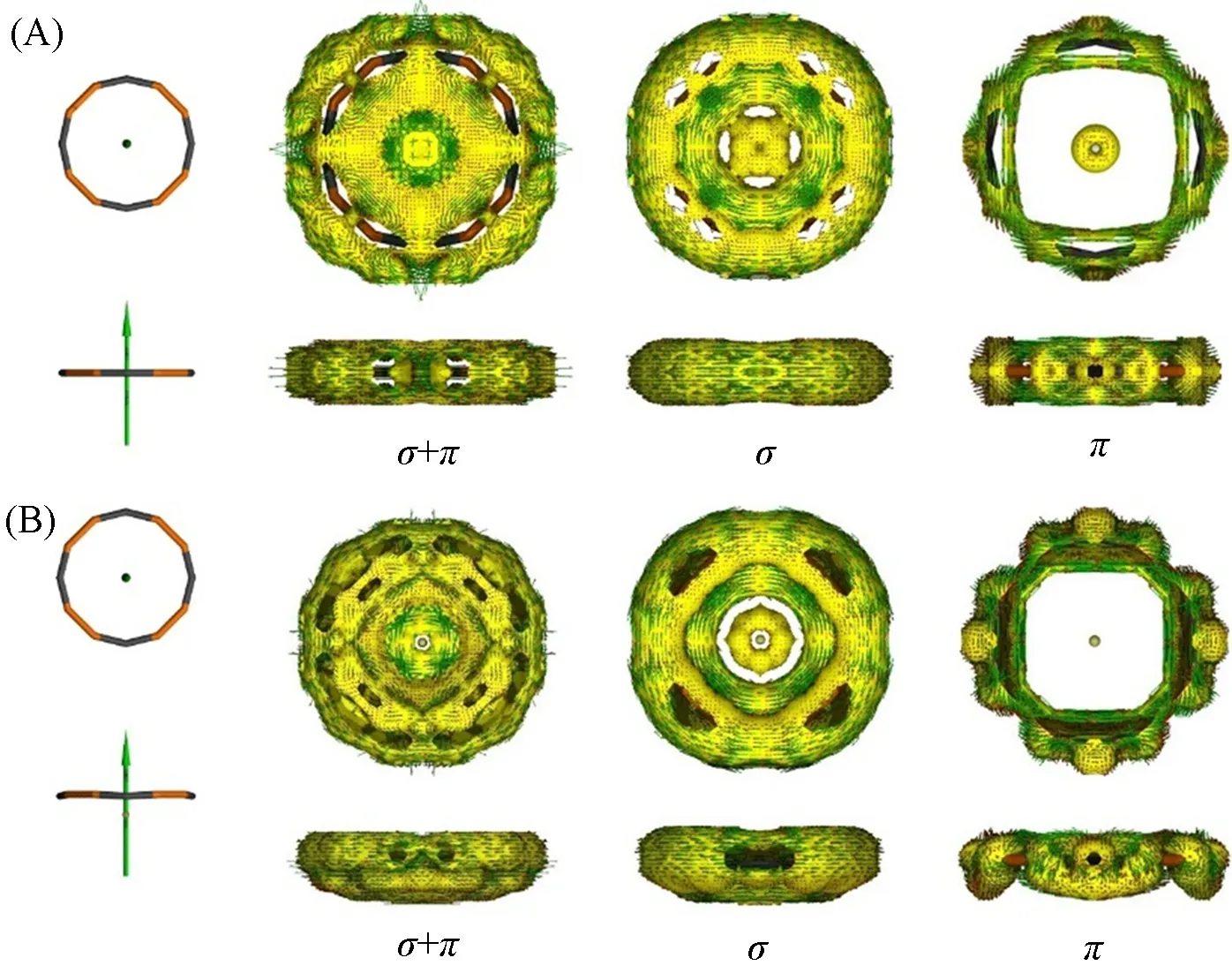
Fig.6 Calculated σ and π-ring current images of CaB8C4(D4h)(A) and SrB8C4(C4v)(B) clusters with an isosurface value of 0.02
在B3LYP/def2-TZVP 水平下, 采用Multiwfn 3.6 软件[39]对轮状CaB8C4团簇进行了QTAIM 分析(图7). 在键临界点(BCP)处, 计算了电子密度(ρ)、 拉普拉斯电子密度(∇2ρ)和能量密度(H). 当ρ>0.2 a.u. 且∇2ρ<0时, 表明具有较大的共价相互作用; 当ρ<0.1 a.u., 并且∇2ρ>0, 表明是离子键.H代表能量密度, 当H<0时, 该键为共价键. 表1列出了轮状CaB8C4团簇在BCPs处的相关参数. 在Ca—C BCPs 处,ρ值为0.0058 a.u., ∇2ρ值为0.032 a.u.,H值为负, 但绝对值非常小, 表明金属和Ca 原子之间存在离子相互作用. 在Ca—B BCPs处,ρ值虽然小于0.1 a.u., 但∇2ρ值为负值(-0.110 a.u.),H值为负, 表明Ca—B之间存在一定的共价作用, 同时也具有一定的离子作用.

Table 1 QTAIM analysis parameters of molecular wheel CaB8C4 cluster
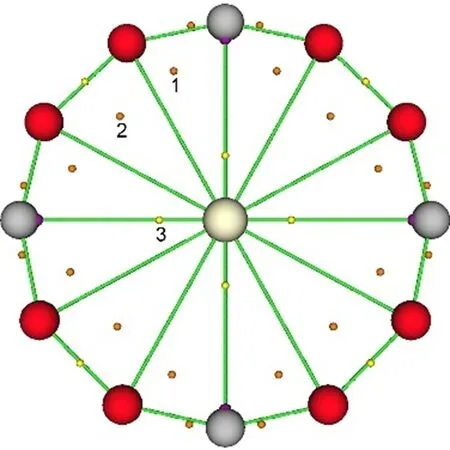
Fig.7 QTAIM analysis of CaB8C4 molecular wheel at B3LYP/def2-TZVP level
大量研究表明, 轮状结构的稳定性得益于中心金属与外环尺寸的匹配效应和双重芳香性的成键特征. 为此, Wang 等[19~21]总结了稳定轮状硼团簇的电子计数规则:n+x+q=K. 电子计数规则适用于以过渡金属为中心的硼分子轮. 而对于本文中的硼碳分子轮, 每个C原子能提供2个电子. 因此, 为了更好地适用于硼碳体系, 需要对这一电子计数规则进行适当调整, 为m+2n+x+q=K(其中,m为B 原子的数目;n为C原子的数目;x为中心金属的价电子;q为团簇所带的电荷数). 为了满足双重芳香的成键特征,K值一般为12 或16. 这一电子计数规则也同样适用于最近报道的十二配位分子轮LaB8C4+/0/-和YB8C4团簇. 值得注意的是, CaB8C4团簇的基态结构虽然是高配位轮状结构, 但其具有10σ/8π冲突芳香性. 归根结底, 该结构稳定性的根本原因可追溯到正交的8π电子. 这4个π轨道正好能稳定4个BCB 单元, 进而提高B8C4环的稳定性. 因此, CaB8C4团簇可作为双重芳香高配位分子轮结构的一个反例.
2.3 拉长状SrB8C4团簇的成键分析
拉长状八配位结构为BaB8C4团簇的基态结构, 同时也是SrB8C4团簇基态结构的共存异构体, 因此有必要阐述该结构的成键特征. 由于两者结构相似, 仅以SrB8C4团簇为例, 对其进行分子轨道分析和AdNDP成键分析. 图8展示了拉长状SrB8C4团簇的占据轨道. 根据原子轨道的类型, 可将这些轨道划分为3 个亚组. 图8(A)中的亚组是基于B 2s/2p和C 2s/2p形成的, 代表外围B7C4环的骨架结构. 这11个轨道遵循轨道构造原则, 从下到上节面数依次为0到5, 形成一套完整体系. 经轨道重组可定域为外围B7C4环的2c-2e B—B/B—Cσ键. 图8(B)中的亚组为5 个离域的σ键, 包括两对近简并轨道HOMO-1/HOMO-3 和HOMO-7/HOMO-5. 图8(C)中的亚组包含5 个离域π轨道, 与图8(B)中的亚组呈现一一对应的关系. 此外, 轨道成分分析表明, 在HOMO, HOMO-1, HOMO-2, HOMO-3, HOMO-4,HOMO-5 和HOMO-6 轨道中存在Sr 4d轨道成分, 表明Sr 的4d轨道和B 2p/C 2p轨道相互作用, 暗示Sr原子具有一定过渡金属性质.

Fig.8 Occupied CMOs of elongated SrB8C4 cluster
图S3(见本文支持信息)更直观地描绘了拉长状SrB8C4团簇的AdNDP 成键结果. 在此结构中有11个2c-2eσ键分布于外围骨架的B—B/B—C 之间. 根据其成键特征, 可以将5 个离域σ键定域为4 个3c-2eσ键和1个5c-2eσ键. 其中前者电子云主要集中在BCB单元, 而后者主要覆盖在右半环的B5单元, 且占据值均大于1.80 |e|. 类似地, 5个离域π键的空间模式与离域σ键完全一致, 且离域π键的占据值略小于σ键, 暗示Sr原子与外围硼碳环之间存在一定的共价作用. 综上所述, 该体系共有42个价电子, 其中, 11对电子用于构建外围硼碳环的骨架结构. 10个离域σ电子和10个离域π电子进一步加强外围硼碳环的稳定性.
2.4 键解离能的分析
为了评估轮状结构中心金属和外围B8C4环之间的相互作用及其热力学稳定性, 根据下列公式在B3LYP-D3/def2-TZVP水平上计算了轮状CaB8C4和SrB8C4团簇的键解离能和固有相互作用能:
上式中B8C4(LM)具有正方形结构, 是势能面上真正的极小结构. 相反, B8C4(frozen)是冻结基态结构后的B8C4环. 计算结果表明, CaB8C4和SrB8C4团簇的键解离能具有较高的负值, 分别为-426.68 和-402.17 kJ/mol, 且固有相互作用能(-543.21和-517.23 kJ/mol)明显高于键解离能, 表明轮状结构具有较高热力学稳定性. 这两体系的固有相互作用能与键解离能之差分别为116.53 和115.06 kJ/mol, 是B8C4单元从四边形转变成圆环结构的异构化能.
2.5 模拟光谱分析
红外光谱与理论化学相结合是表征团簇性质的一种有效方法, 目前已被广泛应用于过渡金属团簇的实验研究[46]. 由于B3LYP方法不适合模拟光谱, 与实验相比偏差较大. 因此, 在M06-2X/def2-TZVP水平上对轮状CaB8C4和拉长状SrB8C4团簇的红外光谱进行模拟(图9). 由图9(A)可知, 轮状CaB8C4团簇的红外光谱分别在323和1386 cm-1处存在两个主要的特征峰, 其中, 最强特征峰在1386 cm-1处, 归属为B—C伸缩振动. 由图9(B)发现, 拉长状八配位SrB8C4团簇的红外光谱有4个较强特征峰, 分别在220, 873, 1609和1731 cm-1处, 最强特征峰在1731 cm-1处. 这些特征峰多归属为伸缩振动.

Fig.9 Infrared spectra of CaB8C4(D4h) molecular wheel(A) and elongated SrB8C4(Cs)(B) clusters
3 结 论
采用密度泛函理论对一系列碱土金属(Ca, Sr, Ba)掺杂硼碳团簇的几何结构和化学成键进行量子化学理论研究, 探索了高配位分子轮的结构、 成键特征和芳香性. 结果表明, 以Ca为中心的十二配位分子轮是CaB8C4团簇的全局极小结构. 随着掺杂原子尺寸的增加, 拉长状八配位结构和轮状结构在能量上存在激烈竞争, 可能共存于SrB8C4团簇的异构体中. 而BaB8C4团簇的基态结构为拉长状八配位结构. NBO电荷分析表明, 团簇内部存在明显的电荷转移, 且中心碱土金属与外围B8C4环之间主要以离子键作用为主, 同时存在微弱共价作用. 化学成键分析揭示十二配位分子轮MB8C4(M=Ca, Sr, Ba)团簇具有8π/10σ电子离域, 是一个冲突芳香性体系. 该体系可视为平面高配位分子轮结构的一个特例.磁感应环电流图和电子定域分析进一步支持分子轮结构的成键结果, 表明离域10σ电子和正交的8π电子有利于提高轮状结构的稳定性. 轨道成分分析揭示碱土金属利用其nd轨道参与部分成键, 表现出过渡金属原子的成键特征. 此外, 也对拉长状SrB8C4团簇的化学成键进行了分析, 证实其存在10σ/10π电子离域. 红外光谱模拟为此类团簇的实验表征提供了一定的理论指导. 这些碱土金属掺杂的硼团簇体系进一步丰富了平面多配位家族.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20230281.
猜你喜欢
辽宁科技大学学报(2022年5期)2023-01-04
山西大学学报(自然科学版)(2022年5期)2022-11-23
大学化学(2021年8期)2021-09-26
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
山东化工(2020年5期)2020-04-07
原子与分子物理学报(2020年5期)2020-03-17
当代陕西(2019年6期)2019-04-17
考试周刊(2018年39期)2018-04-19
厦门大学学报(自然科学版)(2014年2期)2014-08-06
无机化学学报(2014年4期)2014-02-28
