
CRISPR-Cas9介导的基因组编辑技术的研究进展
2015-02-21 06:06郑小梅张晓立于建东孙际宾
生物技术进展 2015年1期
郑小梅, 张晓立, 于建东, 郑 平, 孙际宾*
1.中国科学院系统微生物工程重点实验室, 天津 300308;2.中国科学院天津工业生物技术研究所, 天津 300308;3.天津科技大学生物工程学院, 天津 300457
进展评述
CRISPR-Cas9介导的基因组编辑技术的研究进展
郑小梅1,2, 张晓立1,3, 于建东1,2, 郑 平1,2, 孙际宾1,2*
1.中国科学院系统微生物工程重点实验室, 天津 300308;2.中国科学院天津工业生物技术研究所, 天津 300308;3.天津科技大学生物工程学院, 天津 300457
CRISPR-Cas (clustered regularly interspaced short palindromic repeats-CRISPR-associated proteins)系统为细菌与古生菌中抵御外源病毒或质粒DNA入侵的获得性免疫系统。该系统在crRNA的指导下,使核酸酶Cas识别并降解外源DNA。其中,Ⅱ型CRISPR-Cas系统最为简单,仅包括一个核酸酶Cas9与tracrRNA:crRNA二聚体便可完成其生物功能。基于CRISPR-Cas9的基因组编辑技术的核心为将tracrRNA:crRNA设计为引导RNA,在引导RNA的指导下Cas9定位于特定DNA序列上,进行DNA双链切割,实现基因组的定向编辑。CRISPR-Cas9系统以设计操纵简便、编辑高效与通用性广等优势成为新一代基因组编辑技术,为基因组定向改造调控与应用等带来突破性革命。从CRISPR-Cas9介导的基因组编辑技术的发展与应用等方面综述其最新研究进展,并着重介绍该技术的关键影响因素,为相关研究者提供参考。
CRISPR-Cas系统;Cas9;基因组编辑技术
近年来,基因组编辑技术(genome engineering technologies)的快速发展为生物学研究带来了新纪元。与传统的基因克隆技术不同,基因组编辑技术可直接在基因组上进行DNA序列的敲除、插入、定点突变以及组合编辑等,实现基因功能与调控元件的系统研究,在工业生物工程等方面具有广阔的应用前景。早期,基因组编辑技术主要利用同源重组介导的打靶技术,但由于效率较低(10-6~10-9)[1],极大地限制了其应用。
为解决这一难题,一系列人工核酸内切酶介导的基因组编辑技术被开发,可通过在基因组特定位置上形成DNA双链断裂(DNA double stranded break,DSB),借助于细胞自身的修复系统如非同源末端连接(non-homologous end joining,NHRJ )或同源重组(homology directed repair,HDR),从而实现在不同生物与细胞类型中有效的定点基因组编辑[2]。目前,主要有4种不同的人工核酸内切酶已应用于基因组编辑:巨核酶技术(Meganucleases)[3]、锌指核酸内切酶 (zinc finger endonuclease,ZFN)[4,5]、类转录激活因子效应物核酸酶(transcription activator-like effector nuclease,TALEN)[6~9]与RNA靶向DNA内切酶Cas9[10~12]。
Meganuclease、ZFN与TALEN均可通过蛋白-DNA相互作用识别基因组上的特定DNA序列。Meganuclease具有核酸内切酶结构域与DNA结合域,而ZFN与TALEN分别具有可识别3个与1个核苷酸的DNA结合域,需与核酸内切酶FokI的内切酶结构域形成融合蛋白后,完成基因组特定位点的切割。但这些技术各有不足,如Meganuclease的氨基酸残基与DNA靶序列之间无明确特异性;ZFN的DNA结合域则会受DNA靶序列上下游的影响[13,14],需通过构建不同的融合蛋白来定位到不同的DNA序列,构建操作较为繁琐,成本较高,脱靶风险较高。
RNA靶向内切酶CRISPR-Cas9介导的基因组编辑技术则通过一段短的引导RNA(guide RNA)来识别特定的DNA序列,只需通过改变这段引导RNA序列即可使Cas9定位到新的DNA序列。该技术已被应用于多种生物,包括人、小鼠、大鼠、斑马鱼、秀丽隐杆线虫、植物及细菌。其技术也不断被拓展,如利用多个引导RNA序列可同时进行基因组上多个不同位点的编辑;将其DNA结合域与不同转录调控蛋白相融合,可实现对特定基因的转录激活或抑制[15,16];将对特定序列的调控蛋白模块与基因组或表观基因组的修饰酶相融合,则可实现对基因组的动态控制[15]。CRISPR-Cas9介导的基因组编辑技术以其操作简单、特异性高的特点成为研究热点。
1 CRISPR-Cas系统的发现
CRISPR-Cas9介导的基因组编辑技术的开发与发展基于细菌与古生菌中CRISPR-Cas系统相关的数十年的基础生物学研究。1987年,Ishino等[17]首先在大肠杆菌的碱性磷酸酶基因下游发现被32 bp非重复序列所间隔排列的串联重复序列。随着已测序细菌基因组的增加,发现这种间隔排列的串联重复序列广泛存在于细菌与古生菌中[17]。至2002年,Mojica与Jansen等[18,19]将间隔排列的串联重复序列命名为串联间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)。并且,鉴定出了与CRISPR序列位于同一基因簇的CRISPR相关蛋白Cas,以此将CRISPR基因簇分为3个不同的类型(Ⅰ型、Ⅱ型与Ⅲ型),但其生物学功能还处于未知状态。直至2005年,研究发现间隔序列来源于外源入侵的噬菌体或质粒的DNA序列[20~22],推测CRISPR-Cas系统可能与细菌抵抗外源遗传物质入侵的免疫系统有关[20]。
2007年,Barrangou等[23]首次用实验证明了嗜热链球菌(Streptococcusthermophilus)Ⅱ型CRISPR-Cas系统作为获得性免疫系统的生物功能,揭示出间隔序列指导外源DNA的识别,Cas蛋白负责间隔序列的获得与外源噬菌体的降解 。随后,一系列研究不断揭示出了3个不同类型的CRISPR-Cas系统的具体免疫机制[24~27]。2010年,随着研究的不断深入,发现Ⅱ型CRISPR-Cas系统较为简单,仅需要核酸酶Cas9、成熟的crRNA(CRISPR-derived RNA)与tracrRNA(trans-activating RNA)就可启动对特定外源DNA序列的切割。伴随人工核酸酶ZFN与TALEN介导的基因组编辑技术的迅速发展,许多研究组开始将CRISPR-Cas9开发为RNA靶向的基因组编辑系统,并已在不同的原核与真核生物中获得成功。最近几年,CRISPR-Cas9介导的基因组编辑技术发展更为迅速,在动物建模、作物育种、基因治疗、药物开发以及工业生物工程等不同领域中得到广泛应用。CRISPR-Cas9系统基础研究的深入将推动其介导的基因组规模的精确编辑与调控技术的迅猛发展。
2 CRISPR-Cas系统的免疫机制
CRISPR基因簇由CRISPR序列与Cas蛋白的编码基因组成,CRISPR序列为一系列由不同的间隔序列(spacers)间隔的同向重复序列,间隔序列来源于外源基因片段即原间隔序列(protospacers),如图1(彩图见图版一)所示。Cas蛋白的编码基因将表达为核酸酶、解旋酶与聚合酶等,CRISPR序列则转录加工为短的CRISPR RNAs(crRNAs),指导Cas蛋白与外源入侵片段的识别与降解。
与限制修饰系统相似,CRISPR-Cas系统可识别并降解外源入侵的DNA序列,如图1所示。该过程主要包括四个阶段:首先,噬菌体或质粒的外源DNA进入细胞,即第一阶段噬菌体侵染(phage infection),然后为第二阶段新的间隔序列的获得(spacer acquisition),由特定的Cas蛋白从外源DNA序列 (protospacer)获得间隔序列,并将其整合到细菌基因组的CRISPR位点上。这些间隔序列被同向重复序列所分隔,以确保CRISPR系统对自身序列与外源序列的识别。然后进入第三阶段crRNA的合成与加工(crRNA biogenesis and processing),CRISPR转录为pre-crRNA初级转录本,然后经不同的加工过程形成成熟crRNA(由一个间隔序列与部分重复序列组成),最后通过不同机制指导Cas核酸内切酶对外源DNA序列的识别与降解(target degradation)。
根据参与蛋白与识别机制的不同,CRISPR-Cas系统可分为3个不同的类型:Ⅰ型、Ⅱ型与Ⅲ型CRISPR-Cas系统。在Ⅰ型与Ⅲ型CRISPR-Cas系统中,pre-crRNA初级转录本将被CRISPR相关RNA酶切割重复序列,释放小的成熟crRNA。成熟crRNA将与相关蛋白结合形成RNA-蛋白复合体指导核酸内切酶对外源DNA序列的识别与降解。在Ⅰ型CRISPR-Cas系统中,多个Cas相关蛋白与成熟crRNA形成CRISPR 相关的病毒防御复合体 (CRISPR associated complex for antivirus defense,CASCAD),复合体CASCAD内的crRNA与外源DNA的互补链配对形成R环结构。核酸酶Cas3识别R环结构后,先将DNA的互补链切开,再在其解旋酶和核酸酶作用下将非互补链切开。在Ⅲ型CRISPR-Cas系统中,成熟crRNA可与Csm或Cmr复合体结合,分别对外源DNA或其转录后的RNA序列进行识别降解。
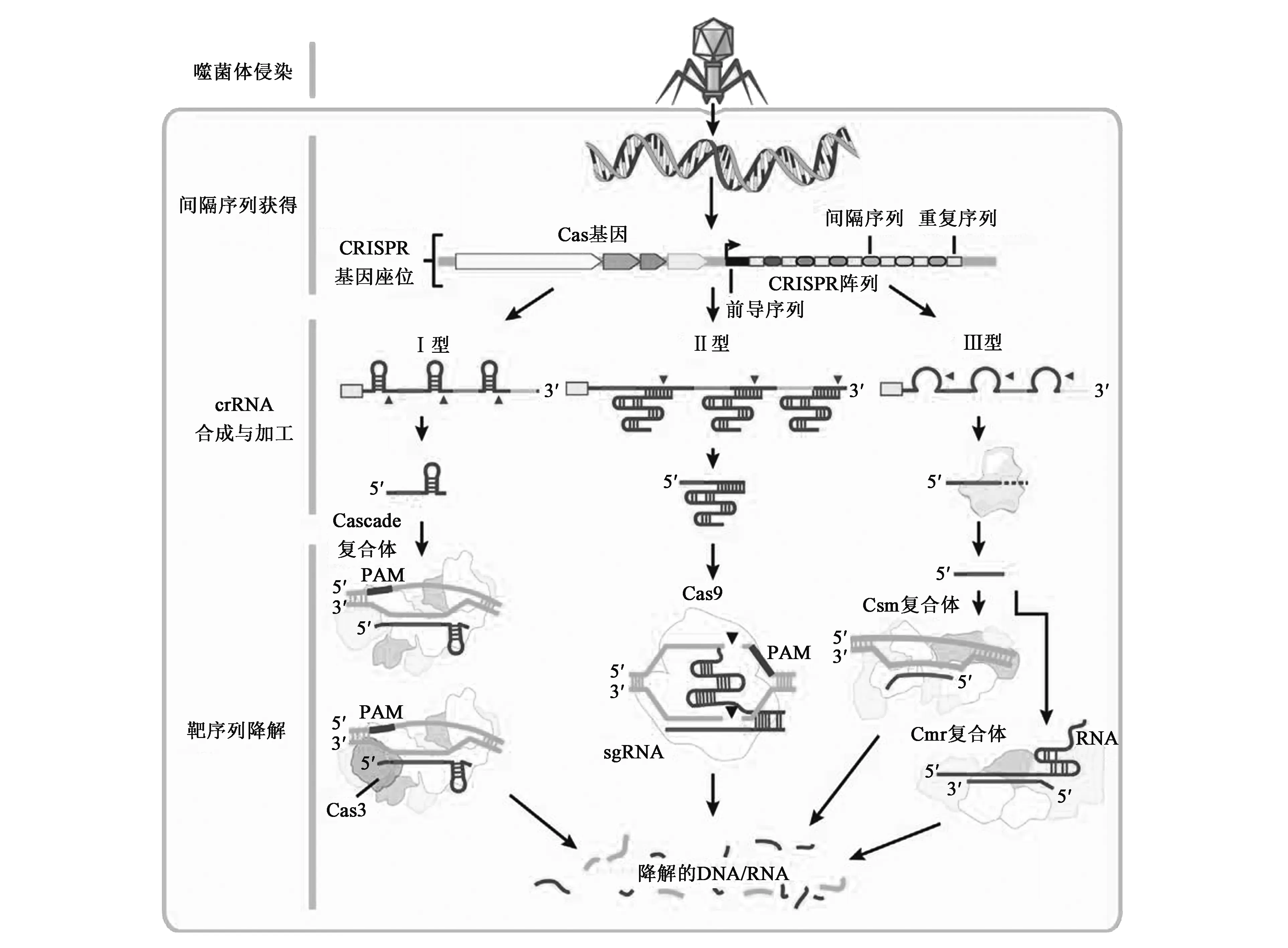
图1 CRISPR-Cas系统抵御外源噬菌体的获得性免疫机制[30]Fig.1 Mechanisms of microbial CRISPR systems in adaptive immunity[30].(彩图见图版一)
在Ⅱ型CRISPR-Cas系统中,还有一种反式激活CRISPR-RNA (tracrRNA)可结合于同向重复序列而形成RNA二聚体,再由RNA内切酶RNase Ⅲ及其他核酸酶进行切割。tracrRNA:crRNA复合体形成的引导RNA与Cas9形成RNA-蛋白复合体后,将结合并扫描外源DNA。当crRNA的间隔序列与外源DNA序列可互补配对时,Cas9则将在互补配对区对外源DNA进行切割。与Ⅰ型与Ⅲ型CRISPR-Cas系统中需要多个蛋白不同,在Ⅱ型CRISPR-Cas系统中仅需要一个核酸内切酶Cas9即可。
在外源DNA序列的原间隔序列(protospacer)的下游存在一个保守的基序(protospacer adjacent motifs,PAMs) 参与Cas蛋白对外源DNA的识别[28]。在Ⅰ型与Ⅱ型CRISPR-Cas系统中,在自身基因组CRISPR序列的下游无PAM (protospacer adjacent motifs)序列,从而将自身基因组DNA序列与外源DNA序列区分开,避免自我免疫。在Ⅲ型CRISPR-Cas系统中,则通过crRNA的5′末端与外源DNA序列的错配来识别并区分外源DNA与自身DNA序列[29]。
3 Ⅱ型CRISPR-Cas9系统
3.1 Cas9的蛋白结构
Cas9的蛋白结构包括α-螺旋组成的识别区(REC)、由HNH结构域与RuvC结构域组成的核酸酶区以及位于C端的PAM结合区(图2,彩图见图版一)。HNH为单个结构域,而 RuvC则分为3个亚结构域:RuvCⅠ位于蛋白的N端,RuvCⅡ/Ⅲ分别位于HNH结构域的两侧。RuvC与HNH可分别对gRNA的DNA互补链与非互补链进行切割,产生平末端的DNA双链断裂。位于RuvC结构域中的D10A突变体可导致RuvC结构域的失活,位于HNH结构域的H840A突变体可导致HNH结构域的失活,但单点突变体可使Cas9成为切口酶(nickase),形成单链DNA断裂。在REC识别区中的一个富含精氨酸的α-螺旋负责与RNA-DNA异源二聚体的3′端8~12个核苷酸的结合。
近年来,对嗜热链球菌SpCas9蛋白结构的研究表明,游离状态下的SpCas9蛋白处于一种自我抑制的构象,HNH结构域的活性位点被RuvC结构域所封闭且远离REC识别区,因而游离的SpCas9不能结合与切割DNA序列。tracrRNA:crRNA可作为一个骨架指导SpCas9蛋白构象的改变,形成一个中心通道可容纳RNA-DNA[31]。
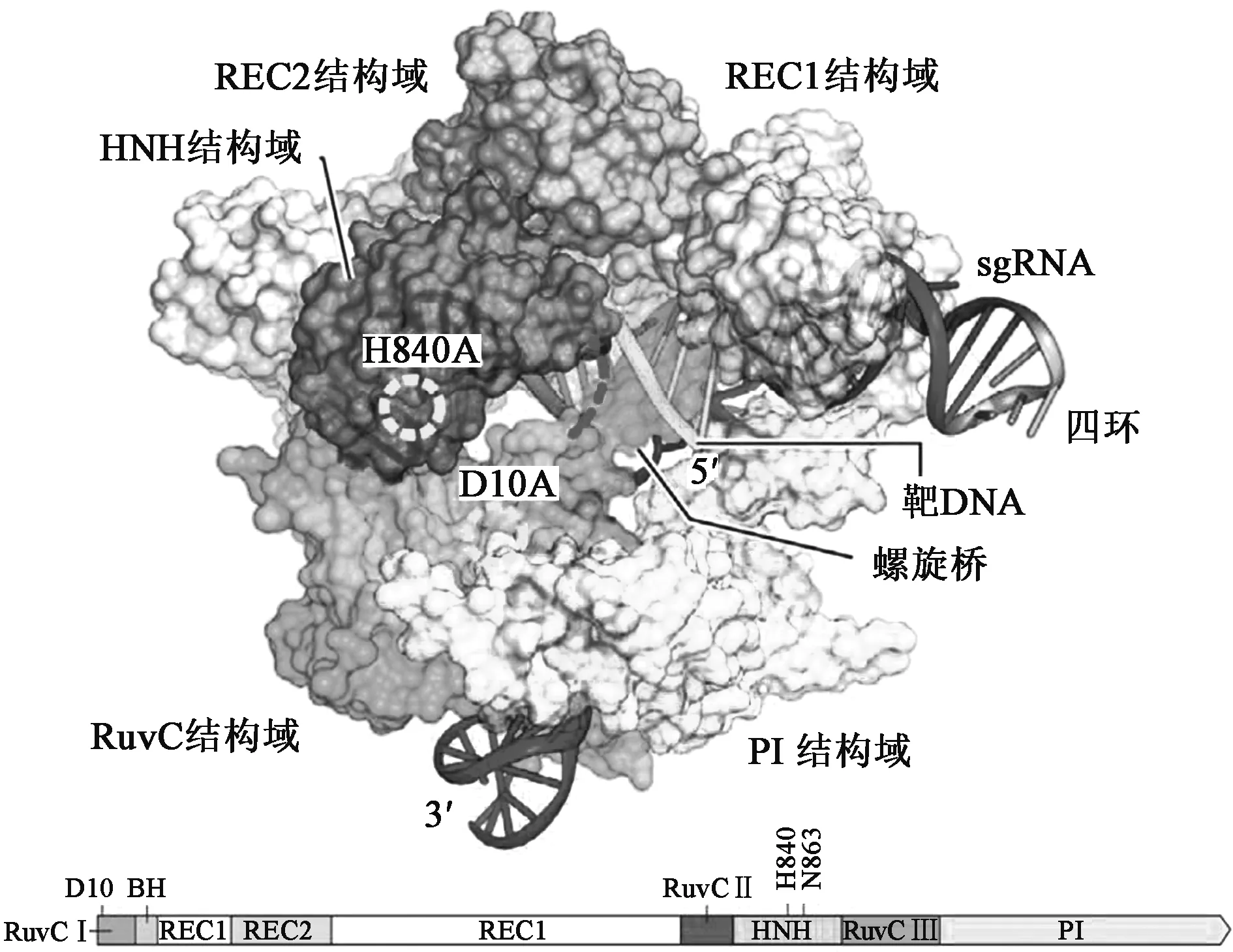
图2 Cas9与引导RNA及靶DNA复合体的晶体结构[30]Fig.2 Crystal structure of S. pyogenes Cas9 in complex with guide RNA and target DNA[30].(彩图见图版一)
3.2 Cas9的识别机制
Cas9与靶DNA的识别依赖于tracrRNA:crRNA复合体以及位于靶位点下游的PAM序列。首先RNA-Cas9复合体沿外源入侵DNA进行扫描,当遇到PAM序列且DNA序列可与crRNA互补配对形成一个R环时,Cas9蛋白将分别利用HNH与RuvC结构域对DNA的互补链与非互补链进行切割,而形成DNA的双链断裂。PAM序列是Cas9识别靶DNA的重要影响因素。SpCas9的研究表明PAM序列的识别将引发Cas9从结合构象向切割构象的转变[30~32]。在嗜热链球菌中,PAM序列多数为5′-NGG,而5′-NAG虽然效率低一些,但也可用于靶DNA的定位,可扩展在基因组编辑中靶DNA的选择范围[33,34]。Cas9的同源蛋白可识别不同的PAM序列,如嗜热链球菌Ⅰ型Cas蛋白识别的PAM序列为5′-NNAGAAW,Ⅲ型Cas蛋白为5′-NGGNG,来源于Neisseriameningitidis的Cas9 识别5′-NNNNGATT PAM序列[28,35,36]。Cas9 蛋白的PAM特异性可进行改造,如将嗜热链球菌Ⅲ型Cas蛋白的PAM识别区替换为StreptococcuspyogenesSpCas9的PAM识别区,则可将其识别的PAM序列由原来的5′-NGGNG变为5′-NGG[30]。
4 CRISPR-Cas9介导的基因组编辑技术
4.1 基本设计原理
CRISPR-Cas9的基因组编辑技术的基本原理为将tracrRNA:crRNA设计为引导RNA,引导RNA包含位于5′端的靶DNA的互补序列以及位于3′-端的tracrRNA:crRNA的类似序列,利用靶DNA的互补序列来定位需编辑的位点,利用tracrRNA:crRNA的类似序列与Cas9结合,如图3(彩图见图版一)所示[37]。该技术仅设计引导RNA就可实现对含有PAM序列的任一靶DNA序列进行敲除、插入与定点突变等修饰。由于设计操作简单、编辑效率高与通用性好等优势,CRISPR-Cas9的基因组编辑技术成为ZFN与TALEN之后的新一代基因组编辑技术。
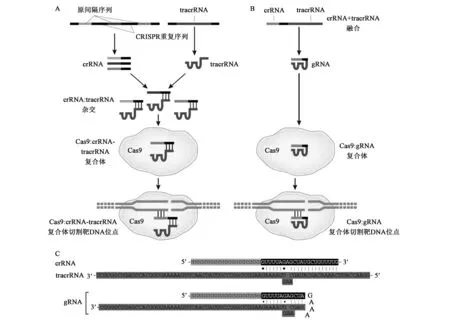
图3 CRISPR-Cas9介导的基因组编辑技术的基本设计原理[37]Fig.3 Naturally occurring and engineered CRISPR-Cas systems[37].A:Ⅱ型CRISPR-Cas系统作用机制示意图,Cas9在tracrRNA:crRNA复合体的引导下,对含有原间隔序列(protospacer)的外源DNA进行切割。B:CRISPR-Cas系统介导的基因组编辑技术的基本原理,通常将crRNA与tracrRNA序列融合为一条引导RNA序列(single gRNA,sgRNA),在sgRNA的引导下,Cas9对位于PAM序列上游,且与sgRNA前20个核苷酸(相当于crRNA)互补的DNA双链进行特异性切割。C:基于tracrRNA:crRNA复合体的gRNA序列设计举例。(彩图见图版一)
CRISPR-Cas9系统还可实现同时对多个不同靶DNA序列的编辑[23,38]。利用CRISPR位点本身的特点,设计对应不同靶DNA序列的间隔序列插入到重复序列之间,经过转录加工后形成多个可定位于不同靶DNA序列的双引导RNA(tracrRNA:crRNA)[10],或者串联不同的sgRNA[11,39]均可对哺乳动物细胞基因组多个不同基因同时进行编辑。
在大多数真核生物中,Cas9所产生的DNA双链断裂被非同源末端接合(non-homologous end joining,NHEJ)或同源重组(homologous recombination,HR)等DNA修复系统修复。但在原核生物中,如肺炎链球菌S.pneumoniae的早期研究中,发现Cas9对基因组的切割是致死的。虽然具体机制尚不清楚,但是这一特点可用于进行细菌基因组编辑后的反筛选。这一策略已在肺炎链球菌S.pneumoniae与E.coli中实现无筛选标记的基因组编辑[33]。将Cas9、引导RNA以及一条含有突变位点的靶DNA的同源重组修复模板共同转化到目的菌株中。若在Cas9对基因组靶DNA序列产生DNA双链断裂后,可利用导入的同源重组修复模板进行DNA修复,由于修复模板在识别互补区或PAM位点中存在突变位点而不能被Cas9再次切割,因而可存活;而未能进行同源重组修复的基因组则由于Cas9的切割降解,无法存活。利用该方法可明显提高基因组编辑后的筛选效率,且在基因组中不残留筛选标记。
4.2 关键影响因素
CRISPR-Cas9基因组编辑技术的迅速发展使越来越多的研究者开始在不同物种中来尝试使用该技术进行基因组的修饰与改造。而在具体实验设计中,必须充分了解该技术的关键影响因素,才能更好应用该技术。
4.2.1 引导gRNA的选择 引导gRNA的设计对于Cas9的活性与效率具有显著的影响。目前,大多数研究将与靶DNA互补的crRNA与tracrRNA融合为一条单独的引导RNA (single guide RNA, sgRNA)。在早期研究中,曾将crRNA与tracrRNA单独表达,设计为双引导RNA (dual guide RNA)[10];但结果表明,与sgRNA相比,双引导RNA更倾向于形成由易错修复系统NHEJ介导的Indel突变。另外,可能由于Cas9:sgRNA两组分的组装比Cas9:crRNA-tracrRNA的组装效率更高,所以sgRNA比双引导RNA具有更快的编辑速度。
sgRNA中所含有的tracrRNA长度的设计也是需考虑的因素之一。在人类基因组编辑过程中,虽然利用含有长至第48位tracrRNA的sgRNA(+48)可在体外对靶DNA进行有效切割[40];但在体内,含有更长tracrRNA 3′端的sgRNA(+72)与sgRNA(+84)对有效的基因组编辑是必须的[11,34]。增加的茎环结构可提高sgRNA的稳定性,有利于Cas9:sgRNA-DNA三聚体正确构象的形成[30]。目前,大多数研究将sgRNA设计为100 nt左右,包含位于5′端20 nt的DNA互补区、crRNA以及位于3′端70~80 nt的tracrRNA。
4.2.2 靶DNA序列的设计 靶DNA设计时,首先要考虑必须在3′端含有PAM序列,然后还需根据所用启动子的特性进行选择。例如依赖于RNA 聚合酶Ⅲ的U6启动子与T7启动子需要转录起始位点分别为G 与GG。如此,使用这些启动子时,sgRNA靶序列就会限制为GN16~19NGG 或GGN15~18NGG。为减少这种限制,有两种策略:一是不考虑启动子带来的限制,在sgRNA 5′端的第一个或前两个核苷酸为错配序列;二是直接在sgRNA 5′端加上额外的G 或GG。目前研究表明,这两种策略均已可获得有活性的sgRNA,但会影响基因组编辑的效率。后续,还需进一步开展大规模的研究来揭示这些错配sgRNA或加长sgRNA对基因组编辑的效率与特异性的影响。
许多研究组开发了可用于靶序列设计的软件或网站,如ZiFiT Targeter Software (http://zifit.partners.org/),CRISPR Design Tool(http://crispr.mit.edu),http://www. genome-engineering.org 与 http://www.egenome.org,以及开放的数据库Addgene等,可辅助进行用于基因组编辑的靶序列设计[30,37]。
4.2.3 Cas9与gRNA的表达与定位 在设计好合适的sgRNA后,就需要考虑如何将Cas9与sgRNA导入目的生物或细胞系中。目前,已在不同的生物与细胞中建立起Cas9与sgRNA的导入方法。在人工培养的哺乳细胞中,可通过电穿孔(electroporation)、核转染(nucleofection)与脂质体介导的转染等方法将非自主复制的质粒DNA导入细胞中,使Cas9 与sgRNA可进行瞬时表达。慢性病毒载体(lentiviral vectors)也已用于在人类或小鼠细胞中组成型表达Cas9与sgRNA。体外转录的RNA也可直接注射导入斑马鱼、果蝇或小鼠的胚胎细胞中;将纯化的Cas9蛋白与sgRNA复合体直接注射到蛔虫体内。除模式动物与细胞系外,Cas9也通过聚乙二醇(polyethylene glycol,PEG)介导的原生质体转化与农杆菌介导的转化等在许多植物中得以成功应用,如小麦、水稻、高粱、烟草和拟南芥等[30]。
许多研究表明,Cas9 与sgRNA的瞬时表达足以介导有效的基因组编辑。虽然Cas9 与sgRNA的组成型表达可能会提高基因组编辑的效率,但也有可能会增加脱靶效应的几率[30,34]。另外,由于Cas9 蛋白来源于细菌如肺炎链球菌,在真核细胞中表达后,还需定位到细胞核内才能介导基因组编辑。为此,需在Cas9 蛋白N端与(或)C端加上真核细胞的核定位信号(nuclear localization signal,NLS)。
4.2.4 提高打靶特异性的策略 虽然现在RNA指导的Cas9核酸酶的特异性机制尚未完全揭示,但许多研究者已开发多种不同策略以提高其特异性,减少脱靶效应。一种策略为降低sgRNA与 Cas9的浓度,但这一方法的效果还有待讨论。有研究表明该方法可显著降低脱靶突变/打靶突变的比例[34],但也有研究称该方法将同时降低脱靶突变与打靶突变的发生[41]。
第二种策略为利用Cas9的切口酶突变体(D10A突变体或H840A突变体)产生DNA单链断裂。因为与DNA双链断裂相比,DNA单链断裂将诱导保真性更高的碱基切除修复[42]。为增加与靶序列特异互补识别的碱基数,利用两条sgRNA与Cas9切口酶的双切口(double-nicking)的方法被开发出来[12,43]。通过在同一靶序列上产生相隔一定距离的两个切口,可模拟双链断裂介导有效的Indel形成。由于脱靶处的切口可被精确修复,因此双切口方法的特异性与野生型Cas9相比,可提高1 500倍[43]。Cas9核酸酶形成的平末端双链断裂或双切口酶形成的粘性末端双链断裂均可激活NHEJ修复的发生,在导入同源重组修复模板的细胞中,将最终形成Indel突变与同源重组突变的混合。因而,利用一条sgRNA、Cas9切口酶与一条同源重组修复模板共同导入细胞后,将进行更有效、更精确的同源重组修复。同时,长同源臂(同源重组修复模板)的存在将大大降低脱靶重组的发生。
第三种策略为sgRNA的截短与修饰,如截短其3′端相当于tracrRNA的序列、在其5′端增加两个额外的GG以及在截短互补识别序列中5′端的2个或3个碱基均可提高Cas9的特异性。前两种方法会损失一定的基因组编辑效率,第三种方法的效率与利用全长sgRNA一样,并能降低脱靶突变的产生,增加对sgRNA:DNA之间单碱基错配的敏感性[44]。以上不同的方法策略可组合使用,进一步降低脱靶突变的产生。Cas9结构与功能研究及其理性改造或定向进化等将会不断提高Cas9的特异性。
5 CRISPR-Cas9基因组编辑技术的应用
CRISPR-Cas9系统介导的基因组编辑技术在生物工程与生物医学方面有广泛的应用[30],例如:可快速建立基因突变的动物或细胞模型,从而揭示遗传变异或表观遗传变异与生物功能或疾病之间的关系;可作为新的作物育种技术,实现抗极端环境、病虫害以及无外源DNA残留的重要农作物的快速获得;还可在藻类或玉米中直接导入有效的乙醇合成代谢途径,获得可持续生产的低成本生物燃料;还可利用基因组编辑改造细菌细胞工厂,生产大宗化学品与精细化学品如药品前体等。
CRISPR-Cas9系统除可用于简便有效的基因组定向编辑外,还具有非常广阔的应用,如基因组规模的功能筛选、内源基因的转录调控与表观遗传调控以及特定染色体位点的标记等[30]。无核酸酶活性的Cas9(“dead”Cas9,dCas9)为Cas9的D10A与H840A双突变体,使HNH与RuvC核酸酶结构域均失活,虽无核酸酶活性,但可作为RNA靶向的DNA结合域,在RNA的指导下与特定DNA序列结合。在E.coli与人类细胞中,将dCas9直接定位结合于启动子区可有效抑制下游基因的转录[45,46]。
dCas9可作为一个DNA结合域招募不同的效应蛋白至特定的基因组位点。例如,dCas9与不同的效应蛋白如转录激活结构域(VP64或NF-κB的p65亚基)或转录抑制结构域(Krüppel-associated box,KRAB)融合后可在人类细胞与小鼠细胞中调控特定靶基因的转录。与基于TALEN设计的转录因子相比,dCas9融合蛋白所引起的转录变化要小一些,但是通过利用2~10个sgRNA来靶向同一个启动子,使dCas9融合的转录激活结构域可协同作用,从而获得较为显著的转录激活反应[30,37]。dCas9与表观遗传效应蛋白融合后,可人为设计对特定基因位点添加或删除如组蛋白修饰与DNA甲基化等特定的表观遗传标记,以研究表观遗传修饰的生物学功能及其在基因组调控网络中的作用[30,37]。在后续工作中,除考虑潜在的脱靶效应外,还需考虑内源表观遗传蛋白对融合蛋白中效应结构域的干扰。
为研究染色体结构组织在基因功能调控中的作用,可利用dCas9与增强型绿色荧光蛋白(enhanced green fluorecence protein,EGFP)融合形成EGFP-dCas9蛋白,在活细胞内对特定基因位点进行成像,以揭示基因组结构(genome architecture)的动态变化[47]。利用来源不同的Cas9与多个gRNA可同时对多个不同基因组位点进行不同颜色的标记,以揭示复杂染色体的结构与组装。
最后,Cas9活性的可诱导调控可通过将Cas9分裂为两个部分来实现,然后分别与可被小分子或光诱导后二聚化的两个结构域融合,在诱导剂存在的条件下,Cas9的两个部分再重新组装起来,形成有活性的蛋白发挥功能。例如,与可诱导的TALEN的构建相似[14],将Cas9的两个部分分别与细胞凋亡过程中的CIB1与CRY2融合后,通过蓝光激活后可重新组装。利用这种方法进行Cas9的活性调控,实现细胞过程中特定基因的实时调控。
6 展望
CRISPR-Cas9作为细菌最为简单的Ⅱ型获得性免疫系统被成功改造为继ZFN与TALEN之后的新一代基因组编辑技术,为基因组定向改造调控与应用等带来突破性革命。与DNA重组技术类似,CRISPR-Cas9系统以设计操纵简便、编辑高效与通用性广等优势,有望成为被广泛应用的基本分子操纵技术,将对生物学的基础研究与应用产生深远的影响。虽然目前CRISPR-Cas9系统中仍有一系列有待解决与发展的问题,例如如何突破PAM序列的限制、如何建立对Cas9特异性(脱靶效应)的全面评价体系、如何构建不同物种中可通用的Cas9与sgRNA导入与表达系统以及如何更有效的激活同源重组修复等。但是基于CRISPR-Cas9系统的迅猛发展速度,我们可以预见,随着相关基础科学研究的深入,CRISPR-Cas9系统将在基因组水平上的基因改造、转录调控与表观遗传调控等不同层次上得到更广阔的发展与应用。
[1] Capecchi M R. Altering the genome by homologous recom-bination[J]. Science,1989,244(4910):1288-1292.
[2] Hsu P D,Lander E S ,Zhang F. Development and applications of crispr-cas9 for genome engineering[J]. Cell,2014,157(6):1262-1278.
[3] Smith J,Grizot S,Arnould S,etal.. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences[J]. Nucleic Acids Res.,2006,34(22):e149.
[4] Urnov F D,Miller J C,Lee Y L,etal.. Highly efficient endogenous human gene correction using designed zinc-finger nucleases[J]. Nature,2005,435(7042):646-651.
[5] Miller J C, Holmes M C, Wang J,etal.. An improved zinc-finger nuclease architecture for highly specific genome editing[J]. Nat. Biotechnol.,2007,25(7):778-785.
[6] Christian M,Cermak T,Doyle E L,etal.. Targeting DNA double-strand breaks with TAL effector nucleases[J]. Genetics,2010,186 (2):757-761.
[7] Miller J C,Tan S,Qiao G,etal.. A TALE nuclease architecture for efficient genome editing[J]. Nat. Biotechnol.,2011,29(2):143-148.
[8] Boch J,Scholze H,Schornack S,etal.. Breaking the code of DNA binding specificity of TAL-type Ⅲ effectors[J]. Science,2009,326(5959): 1509-1512.
[9] Moscou M J, Bogdanove A J. A simple cipher governs DNA recognition by TAL effectors[J]. Science,2009,326(5959):1501.
[10] Cong L,RanF A,Cox D,etal.. Multiplex genome engineering using CRISPR/Cas systems[J]. Science,339(6121):819-823.
[11] Mali P,Yang L,Esvelt K M,etal.. RNA-guided human genome engineeringviaCas9[J]. Science,2013,339(6121):823-826.
[12] Mali P,Aach J,Stranges P B,etal.. Cas9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering[J]. Nat. Biotechnol.,2013,31(9):833-838.
[13] Maeder M L,Thibodeau-Beganny S,Osiak A,etal.. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification[J]. Mol. Cell,2008,31(2):294-301.
[14] Juillerat A,Dubois G,Valton J,etal.. Comprehensive analysis of the specificity of transcription activator-like effector nucleases[J]. Nucleic Acids Res.,2014,42(8):5390-5402.
[15] Konermann S,Brigham M D,Trevino A E,etal.. Optical control of mammalian endogenous transcription and epigenetic states[J]. Nature,2013,500(7463):472-476.
[16] Mendenhall E M,Williamson K E,Reyon D,etal.. Locus-specific editing of histone modifications at endogenous enhancers[J]. Nat. Biotechnol.,2013,31 (12):1133-1136.
[17] Ishino Y,Shinagawa H,Makino K,etal.. Nucleotide sequence of theiapgene,responsible for alkaline phosphatase isozyme conversion inEscherichiacoli,and identification of the gene product[J]. J. Bacteriol.,1987,169 (12):5429-5433.
[18] Mojica F J,Díez-Villaseor C,Soria E,etal.. Biological significance of a family of regularly spaced repeats in the genomes of Archaea,Bacteria and mitochondria[J]. Mol. Microbiol.,2000,36(1): 244-246.
[19] Jansen R,Embden J D,Gaastra W,etal.. Identification of genes that are associated with DNA repeats in prokaryotes[J]. Mol. Microbiol.,2002,43(6):1565-1575.
[20] Mojica F J,Díez-Villaseor C,García-Martínez J,etal.. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements[J]. J. Mol. Evol.,2005,60(2): 174-182.
[21] Pourcel C,Salvignol G,Vergnaud G. CRISPR elements inYersiniapestisacquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies[J]. Microbiology,2005,151 (Pt 3):653-663.
[22] Bolotin A,Quinquis B,Sorokin A,etal.. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin[J]. Microbiology,2005,151(Pt 8):2551-2561.
[23] Barrangou R,Fremaux C,Deveau H,etal.. CRISPR provides acquired resistance against viruses in prokaryotes[J]. Science,2007,315 (5819):1709-1712.
[24] Brouns S J,Jore M M,Lundgren M,etal.. Small CRISPR RNAs guide antiviral defense in prokaryotes[J]. Science,2008,321(5891):960-964.
[25] Marraffini L A,Sontheimer E J.CRISPR interference limits horizontal gene transfer instaphylococciby targeting DNA[J]. Science,2008,322 (5909):1843-1845.
[26] Hale C R,Zhao P,Olson S,etal.. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex[J]. Cell,2009,139(5): 945-956.
[27] Hale C R,Majumdar S,Elmore J,etal.. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs[J]. Mol. Cell,2012,45(3):292-302.
[28] Deveau H,Barrangou R,Garneau J E,etal.. Phage response to CRISPR-encoded resistance inStreptococcusthermophilus[J]. J. Bacteriol.,2008,190(4):1390-1400.
[29] Marraffini L A,Sontheimer E J. Self versus non-self discrimination during CRISPR RNA-directed immunity[J]. Nature,2010,463(7280):568-571.
[30] Nishimasu H,Ran F A,Hsu P D,etal.. Crystal structure of Cas9 in complex with guide RNA and target DNA[J]. Cell,2014,156(5):935-949.
[31] Jinek M,Jiang F,Taylor D W,etal.. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation[J]. Science,2014,343 (6176):1247997.
[32] Sternberg S H,Redding S,Jinek M,etal.. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9[J]. Nature,2014,507(7490):62-67.
[33] Jiang W,Bikard D,Cox D,etal.. RNA guided editing of bacterial genomes using CRISPR-Cas systems[J]. Nat.Biotechnol.,2013,31(3):233-239.
[34] Hsu P D,Scott D A,Weinstein J A,etal.. DNA targeting specificity of RNA-guided Cas9 nucleases[J]. Nat. Biotechnol.,2013,31(9):827-832.
[35] Horvath P,Romero D A,Coté-Monvoisin A C,etal.. Diversity,activity,and evolution of CRISPR loci inStreptococcusthermophilus[J]. J. Bacteriol.,2008,190(4): 1401-1412.
[36] Zhang Y,Heidrich N,Ampattu B J,etal.. Processing-independent CRISPR RNAs limit natural transformation inNeisseriameningitidis[J]. Mol. Cell,2013,50(4):488-503.
[37] Sander J D,Joung J K. CRISPR-Cas systems for editing,regulating and targeting genomes[J]. Nat. Biotechnol.,32(4):347-355.
[38] Garneau J E,Dupuis M,Villion M,etal.. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA[J]. Nature,2010,468(7320):67-71.
[39] Wang H,Yang H,Shivalila C S,etal.. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering[J]. Cell,2013,153(4):910-918.
[40] Jinek M,Chylinski K,Fonfara I,etal.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity[J]. Science,2012,337(6096):816-821.
[41] Fu Y,Foden J A,Khayter C,etal.. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells[J]. Nat. Biotechnol.,2013,31(9):822-826.
[42] Dianov G L,Hübscher U. Mammalian base excision repair:The forgotten archangel[J]. Nucleic Acids Res.,2013,41(6):3483-3490.
[43] Ran F A,Hsu P D,Lin CY,etal.. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity[J]. Cell,2013,154(6):1380-1389.
[44] Fu Y,Sander J D,Reyon D,etal.. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs[J]. Nat. Biotechnol.,2014,32(3):279-284.
[45] Bikard D,Jiang W,Samai P,etal.. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system[J]. Nucleic Acids Res.,2013,41(15):7429-7437.
[46] Qi L S,Larson M H,Gilbert L A,etal.. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression[J]. Cell,2013,152(5):1173-1183.
[47] Chen B, Gilbert L A, Cimini B A,etal.. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system[J]. Cell,155:1479-1491.
[48] Charpentier E,Marraffini L A. Harnessing CRISPR-Cas9 immunity for genetic engineering[J]. Curr. Opin. Microbiol.,2014, 19:114-119.
CRISPR-Cas9-based Genome Engineering
ZHENG Xiao-mei1,2, ZHANG Xiao-li1,3, YU Jian-dong1,2, ZHENG Ping1,2, SUN Ji-bin1,2*
1.KeyLaboratoryofSystemsMicrobialBiotechnology,ChineseAcademyofSciences,Tianjin300308,China;2.TianjinInstituteofIndustrialBiotechnology,ChineseAcademyofSciences,Tianjin300308,China;3.SchoolofBiologicalEngineering,TianjinUniversityofScienceandTechnology,Tianjin300457,China
CRISPR-Cas (clustered regularly interspaced short palindromic repeats-CRISPR-associated proteins) systems are the adaptive immune system in bacteria and archaea that defends against infectious viruses and plasmids. Immunity is mediated by Cas nucleases, which use small RNA guides (the crRNAs) to specify a cleavage site within the genome of invading nucleic acids. In type Ⅱ CRISPR-Cas systems, the DNA-cleaving activity is performed by a single enzyme Cas9 guided by an RNA duplex. Using synthetic single RNA guides, Cas9 can be reprogrammed to create specific double-stranded DNA breaks in the genomes of a variety of organisms, ranging from human cells to bacteria, and thus constitutes a powerful tool for genetic engineering. In this review, we described the development and applications of CRISPR-Cas9 system with highlighting the practical considerations for implementing CRISPR-Cas9 technology.
CRISPR-Cas system; Cas9; genome engineering
2014-11-04; 接受日期:2014-12-04
国家863计划项目(2013AA020302);国家自然科学基金面上项目(31370113)资助。
郑小梅,助理研究员,博士,从事黑曲霉分子生物学与系统生物学研究。E-mail:zheng_xm@tib.cas.cn。*通信作者:孙际宾,研究员,博士生导师,主要从事微生物代谢工程与系统生物技术研究。Tel:022-84861949;E-mail:sun_jb@tib.cas.cn
10.3969/j.issn.2095-2341.2015.01.01
猜你喜欢
舰船科学技术(2022年11期)2022-07-15
无机化学学报(2020年7期)2020-07-20
中国预防兽医学报(2020年2期)2020-06-01
三农资讯半月报(2020年8期)2020-05-13
西藏农业科技(2019年3期)2019-11-04
广州大学学报(自然科学版)(2019年1期)2019-05-07
现代园艺(2018年3期)2018-02-10
上海农业学报(2017年3期)2017-04-10
中国病理生理杂志(2017年2期)2017-01-17
天津科技大学学报(2016年1期)2016-02-28
