
泮托拉唑钠肠溶胶囊的质量控制及稳定性研究
2016-05-23 02:54尹美艳
安徽医药 2016年4期
尹美艳,高 蓉
(杭州中美华东制药有限公司,浙江 杭州 310000)
泮托拉唑钠肠溶胶囊的质量控制及稳定性研究
尹美艳,高蓉
(杭州中美华东制药有限公司,浙江 杭州310000)
摘要:目的建立泮托拉唑钠肠溶胶囊含量及有关物质的质量控制方法,并对上市品进行稳定性研究。方法以Agilent C(18)柱(250 nm×4.6 nm,5 μm)为色谱柱,流动相为磷酸二氢钠缓冲(1.20 g的磷酸二氢钠溶解于1 000 mL的水中,用磷酸调节pH至7.0)—乙腈(68∶32),流速1 mL·min(-1),检测波长288 nm,对胶囊进行有关物质及含量的检测;采用加速试验,在温度(40±2)℃、相对湿度(75±5)%的条件下放置6个月,考察其稳定性。结果泮托拉唑钠在(1.0~10.0)×10(-3 ) g·L(-1)浓度范围内线性关系良好,检 测限为0.20 ng,定量限为0.60 ng,平均加样回收率为100.57%(RSD=0.41%),测定样品平均含量为100.67%,有关物质均小于0.2%。稳定性试验结果含量在99.70%~101.16%,有关物质小于0.2% 。结论该方法快速,简单,稳定,重现性好,可作为泮托拉唑钠肠溶胶囊的含量及有关物质的检测方法。稳定性检测结果符合要求。
关键词:泮托拉唑钠肠溶胶囊;HPLC法;含量;有关物质;稳定性
泮托拉唑钠是继奥美拉唑钠、兰索拉唑钠之后的为第三代质子泵抑制剂,由德国百克顿公司研制生产,于1994年在南非首次上市。化学名为:5-二氟甲氧基-2-[(3,4-二甲氧基-2-吡啶基)-甲基]亚硫酰基-1H-苯并咪唑钠盐一水合物,分子式为C16H12F2N3NaO4S·H2O。临床主要用于治疗急性消化性溃疡(胃、十二指肠溃疡)、反流性食管炎及卓—艾氏综合征。 通过作用于H+/K+-ATP酶抑制胃酸和组胺分泌、作用时间长、起效快、强度大等优点成为治疗溃疡的一线药物。抗溃疡疗效方面要明显优于雷尼替丁,生物利用度高于奥美拉唑和兰索拉唑[1-2]。泮托拉唑钠肠溶胶囊国外药典未收载,进口注册标准及2015版《中国药典》[3-4]收载了泮托拉唑钠及泮托拉唑钠肠溶胶囊的HPLC的检验方法,陈玉玲等虽报道过有关泮托拉唑钠肠溶胶囊有关物质的检测方法[5-7],通过试验发现上述方法存在分离度低,重现性差等弊端,本文通过对HPLC条件的优化得到重现性好、分离度高、方便快捷的高效液相条件,使得泮托拉唑钠肠溶胶囊质量得到有效控制。
1仪器与试药
Agilent 1260型高效液相色谱仪(DAD),安捷伦色谱工作站,电子分析天平(XSE 105DU)。色谱纯乙腈(Fisher公司),重蒸馏水,磷酸二氢钠(分析纯,国药集团化学试剂有限公司),磷酸(分析纯,国药集团化学试剂有限公司),氢氧化钠(分析纯,国药集团化学试剂有限公司)。泮托拉唑钠对照品(中国食品药品检定研究院,批号100575-201408),泮托拉唑钠肠溶胶囊(山东绿叶制药有限公司,批号14110202,15021001,15031802,规格40 mg)。
2方法和结果
2.1色谱条件色谱柱:Agilent C18柱(250 nm×4.6 nm,5 μm);流动相:磷酸二氢钠缓冲(1.20g的磷酸二氢钠溶解于1 000 mL的水中,用磷酸调节pH至7.0)-乙腈(68∶32);流速:1 mL·min-1;检测波长:288 nm;进样量:20 μL;柱温:40℃。
2.2溶液配制
2.2.1含量测定对照品溶液取泮托拉唑钠对照品适量,加入0.001 mol·L-1氢氧化钠溶液-乙腈(1∶1)混合溶剂溶解并定量稀释至每1 mL含有0.04 mg的溶液,备用。
2.2.2含量测定供试品溶液取泮托拉唑钠肠溶胶囊20粒,取内容物研细,精密称量适量(约相当于泮托拉唑钠40 mg),置100 mL量瓶中,加入0.001 mol·L-1氢氧化钠溶液—乙腈(1∶1)混合溶剂,超声使泮托拉唑钠溶解,补加上述溶剂稀释至刻度,摇匀,取滤液10 mL,置100 mL量瓶中,加上述溶剂稀释至刻度,摇匀,作为供试品溶液备用。
2.2.3 有关物质测定供试品溶液称取“2.2.2”项中泮托拉唑钠肠溶胶囊内容物适量,研细,取粉末精密称定,置50 mL的量瓶中,加入0.001 mol·L-1氢氧化钠溶液—乙腈(1∶1)混合溶剂适量,超声溶解,加入上述溶液稀释至刻度,摇匀。配制成1 mL中含有约0.4 mg泮托拉唑钠的溶液。
2.2.4 有关物质对照溶液精密量取“2.2.3”项中的有关物质供试品溶液1 mL,置200 mL的量瓶中,加入0.001 mol·L-1氢氧化钠溶液—乙腈(1∶1)混合溶剂稀释至刻度,摇匀。
2.2.5 阴性对照溶液按照处方比例称取羟丙甲纤维素(Hydroxypropyl Methyl Cellulose,HPMC )、吐温-80(Tween-80)相当于一粒胶囊的辅料量,置于100 mL量瓶中,加0.001 mol·L-1氢氧化钠溶液—乙腈(1∶1)混合溶剂稀释至刻度,超声,滤过,取1 mL加入上述溶剂稀释与样品相同倍数,即得。
2.3方法学考察
2.3.1专属性试验取“2.2.1”项下泮托拉唑钠对照品溶液精密量取 20 μL进色谱仪,按照上述色谱条件测定,记录色谱图。按照上述方法分别取“2.2.2”项下含量供试品溶液、 “2.2.3”项有关物质供试品溶液、“2.2.4”项有关物质对照及“2.2.5”阴性对照品溶液,稀释至同等浓度,进色谱仪测定,记录色谱图,结果辅料峰未干扰测定(图1)。分别取泮托拉唑钠肠溶胶囊适量,取内容物研细,精密称定粉末(约相当于主药40 mg)进行如下测试:(1)酸破坏:将精密称定后的粉末置具塞试管中,用0.1 mol·L-1盐酸溶液25 mL,摇匀,置于100℃水浴锅中4 h,放冷。加入0.1 mol·L-1氢氧化钠中和并转移至100 mL的容量瓶中,用0.001 mol·L-1氢氧化钠溶液—乙腈(1∶1)混合溶剂稀释至刻度,摇匀,过滤。(2)碱破坏:将精密称定后的粉末置具塞试管中,用0.1 mol·L-1氢氧化钠溶液25 mL,摇匀,置于100℃水浴锅中4 h,放冷。加入0.1 mol·L-1盐酸中和并转移至100 mL的容量瓶中,用0.001 mol·L-1氢氧化钠溶液—乙腈(1∶1)混合溶剂稀释至刻度,摇匀,过滤。(3)氧化破坏:将精密称定后的粉末置具塞试管中,加入30%过氧化氢溶液10 mL,摇匀,放置12 h,加0.001 mol·L-1氢氧化钠溶液—乙腈(1∶1)混合溶剂稀释至刻度,摇匀,过滤。(4)加热破坏:将精密称定后的粉末置具塞试管中,加0.001 mol·L-1氢氧化钠溶液-乙腈(1∶1)混合溶剂稀释,配置成浓度0.04 g·L-1溶液25 mL,置于100℃水浴锅中4 h,放冷,加上述溶液转移至100 mL容量瓶中,用上述溶液稀释至刻度,摇匀,过滤。(5)光破坏:将精密称定后的粉末置具塞试管中,加0.001 mol·L-1氢氧化钠溶液-乙腈(1∶1)混合溶剂稀释,配置成0.04 g·L-1溶液25 mL,置于强光(5 000 Lx)下照射12 h后,放冷。按照上述色谱条件进行测定,记录色谱图。结果显示样品在经过酸、碱、氧化、加热、光照破坏后主成分与各降解产物可以有效分离(图2)。

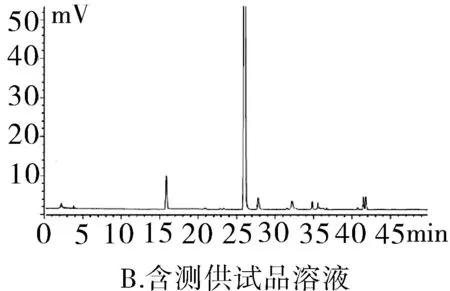
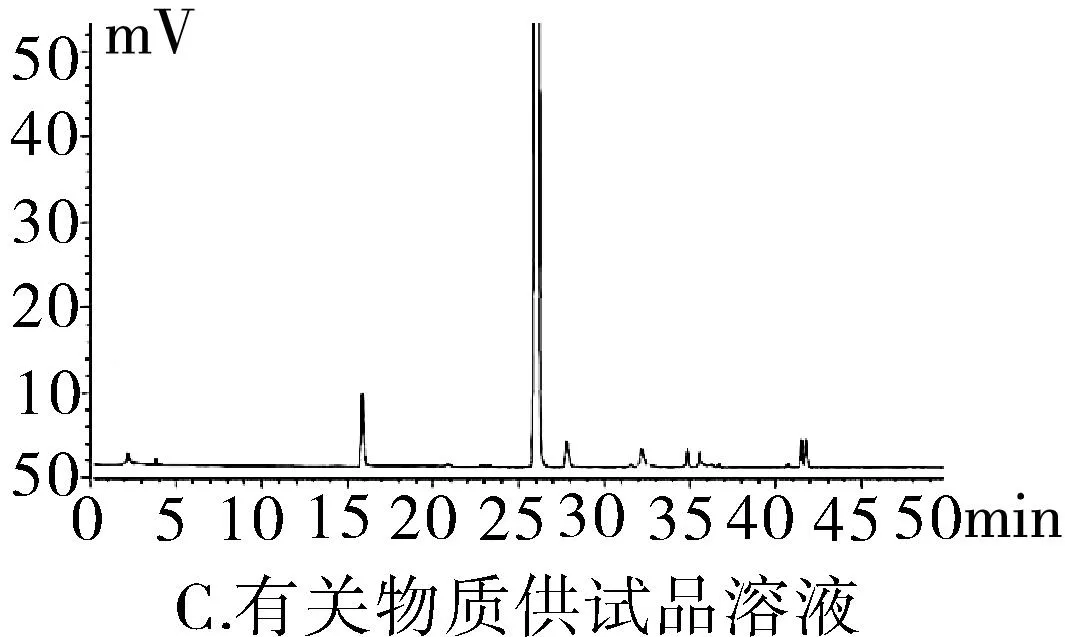
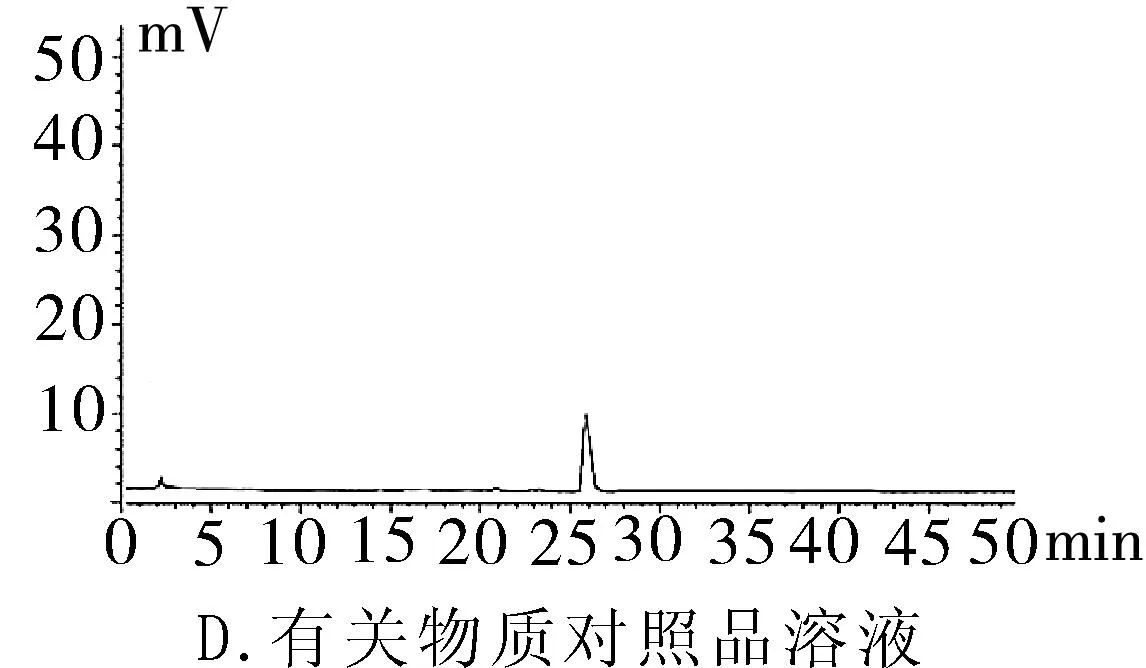

图1 HPLC图谱
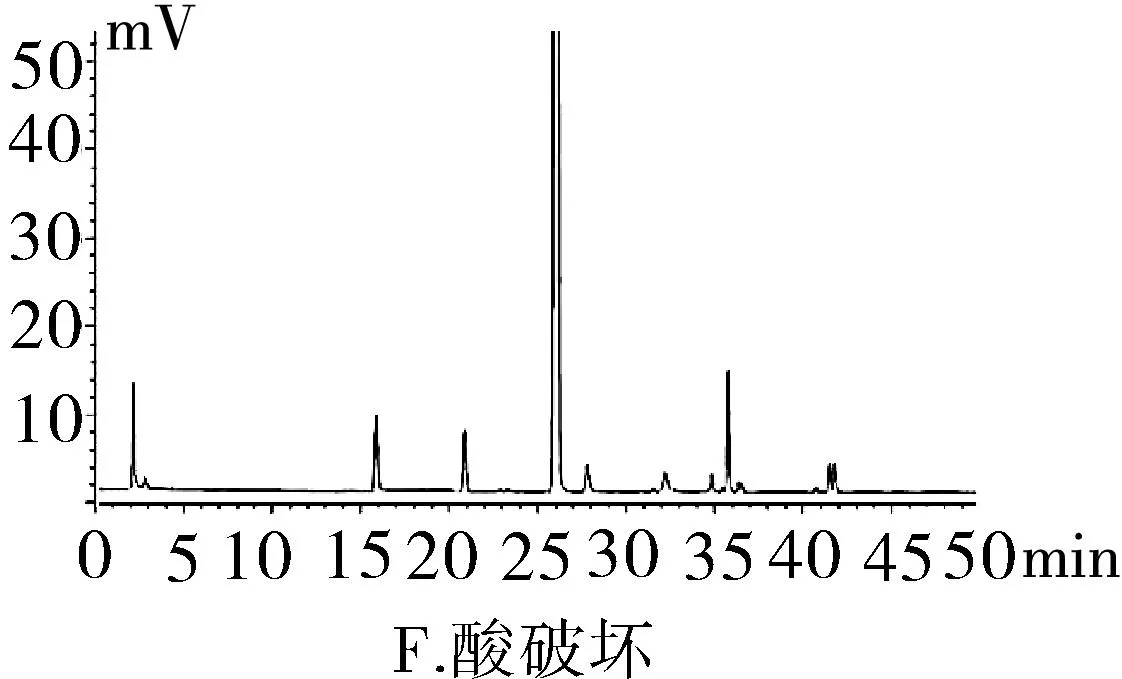
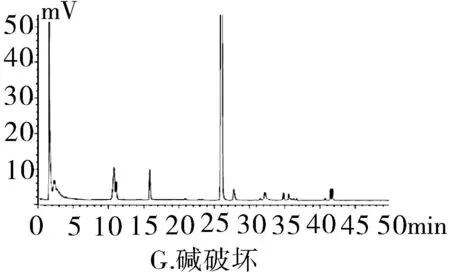



图2 泮托拉唑钠方法学图谱
2.3.2系统适用性试验分别精密量取“2.2.1”项中泮托拉唑钠对照品溶液、“2.2.2”泮托拉唑钠肠溶胶囊供试品溶液及“2.2.5”项阴性对照溶液20 μL,按上述色谱条件进样测定,色谱柱的理论塔板数按照泮托拉唑钠计算不得少于3 000,泮托拉唑峰与相邻杂质峰分离度不低于2.0。
2.3.3检测限及定量限测定取“2.2.1”项中配制的泮托拉唑钠对照品溶液,稀释至0.4×10-3g·L-1,按照上述色谱条件进样,根据信噪比法进行试验,得到的结果:检测限为0.20 ng(S/N=3),定量限为0.60 ng(S/N=10)。
2.3.4精密度及重复性试验取“2.2.1”项下泮托拉唑钠对照品溶液,按照上述色谱条件进样,重复测定6次,记录泮托拉唑钠的峰面积,计算相对标准偏差RSD=0.31%,表明仪器精密度良好。

表1 加样回收率试验
取14110202批次泮托拉唑钠肠溶胶囊6粒,分别取内容物研细,精密称量适量粉末(相当于主药20 mg),加0.001 mol·L-1氢氧化钠溶液—乙腈(1∶1)混合溶剂配制成浓度为0.4 g·L-1供试品溶液6份,按照上述色谱条件在同一高效液相色谱仪上分别进样,记录泮托拉唑钠峰面积,计算相对标准偏差RSD=0.37%,显示重现性良好。
2.3.5加样回收率试验按照处方比例称量适量辅料(相当于1粒胶囊辅料的量),分别加入处方量的80%,100%,120%的泮托拉唑钠的对照品,加0.001 mol·L-1氢氧化钠溶液—乙腈(1∶1)混合溶剂溶解并稀释至刻度,摇匀,每个含量的溶液分别取3份作为供试品溶液。精密量取20 μL溶液,按照上述色谱条件进高效液相色谱仪,记录图谱,结果见表1。计算得平均加样回收率为100.57%,RSD=0.41%。
2.3.6样品溶液稳定性试验取14110202批次样品制备成浓度为0.4 g·L-1供试品溶液,分别在0、2、4、6、12、18 h精密吸取20 μL进样,记录色谱图,根据泮托拉唑钠峰面积,计算其相对标准偏差RSD为0.07%,说明在18 h内泮托拉唑钠在溶液内稳定。
2.4样品测定
2.4.1有关物质测定取3批上市品泮托拉唑钠肠溶胶囊各10粒,取内容物研细,精密称量适量粉末(相当于泮托拉唑钠40 mg),置100 mL的量瓶中,加0.001 mol·L-1氢氧化钠溶液—乙腈(1∶1)混合溶剂振荡溶解,并稀释至刻度,摇匀,过滤,滤液作为供试品。取“2.2.1”项中泮托拉唑钠对照品溶液稀释与供试品同等浓度作为对照溶液。精密量取供试品和对照品溶液20 μL进高效液相色谱仪,记录色谱图。根据面积归一化法计算有关主峰及有关物质的峰面积。
2.4.2含量测定取3批次上市品泮托拉唑钠肠溶胶囊各10粒,取内容物研细,制备成浓度同2.2.1项中浓度作为供试品溶液。分别精密量取“2.2.1”项中泮托拉唑钠对照品溶液稀释与供试品各20 μL,进高效液相色谱仪,记录图谱。按照外标法分别测定测定3批上市品含量。3批样品的有关物质及含量的结果见表2。
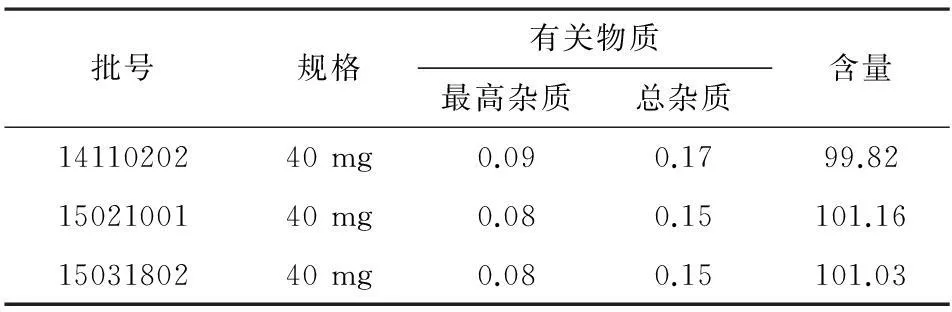
表2 泮托拉唑钠肠溶胶囊有关物质及含量测定结果

表3 泮托拉唑钠肠溶胶囊的稳定性果
2.5稳定性研究参照《中国药典》2015版附录9001 原料药物与制剂稳定性试验指导原则,对市售的3批制剂(14110202,15021001,15031802)进行加速试验,取市售最小包装,在温度(40±2)℃、相对湿度(75±5)%的条件下放置6个月。分别于1月、2月、3月和6月取样一次,按照“2.4”项中有关物质及含量的测定方法对泮托拉唑钠肠溶胶囊的稳定性进行考察,结果见表3。
3讨论
泮托拉唑钠因含有亚磺酰基苯并咪唑机构,导致产品对热、光不稳定,在酸性条件下不稳定[8],在做产品的方法学时,选择0.001 mol·L-1氢氧化钠溶液—乙腈(1∶1)的混合溶液溶解测定,避免在偏酸或中性的溶剂中产生降解杂质。
本文通过对比《中国药典》提供的Kromasil、Waters及Agilent的C18柱,通过在同一仪器上的验证得到Agilent的C18柱具有分离度高、柱效稳定,能有效分离杂质优点,并且在288 nm,主峰和破坏杂质峰有最大吸收。通过对泮托拉唑钠肠溶胶囊的测定结果,在拟定的HPLC条件下,主峰、辅料峰及有关物质能够达到基线分离,符合检测要求。方便简便,快捷,且能满足质量控制要求。
通过对3批不同批次的市售药品进行了6个月加速试验,从测定结果可以看出,产品稳定性较好,在制剂的条件下能有效避免原料药不稳定的因素,说明产品的制剂工艺成熟、稳定,能有效控制杂质,保证含量,对药物在临床的使用提供安全保障。
参考文献:
[1]郁心圃. 泮托拉唑钠和奥美拉唑治疗胃溃疡的对照研究[J].实用临床医药杂志,2012,16(19):97-98.
[2]张俊平.奥美拉唑、兰索拉唑、泮托拉唑治疗胃溃疡临床疗效观察[J].现代诊断与治疗,2015,26(4):800-801.
[3]国家食品药品监督管理局.进口药品注册标准JX20070162[S].
[4]国家药典委员会.中国药典(二部)[S].北京:中国医药科技出版社,2015:705.
[5]陈玉玲.HPLC法测定泮托拉唑钠肠溶微丸胶囊的含量[J].科技创新与应用,2012(1):38.
[6]马力,黄良升,张玉.高效液相色谱法测定泮托拉唑钠肠溶微丸胶囊的含量[J].医学导报,2009,28(7):924-925.
[7]张舒,张昀.HPLC梯度洗脱法测定泮托拉唑钠肠溶胶囊的有关物质[J].中国药师,2015,18(6):943-945.
[8]王婧思,李桂龙,王成港,等.泮托拉唑钠有关物质分析方法的比较[J].药物评价研究,2012,35(2):109-112.
The study of quality and stability on pantoprazole sodium enteric-coated capsules
YIN Mei-yan,GAO Rong
(HuadongMedicineCo.,LTD.,Hangzhou,Zhejiang310000)
Abstract:ObjectiveAn HPLC method was established for determination of the content of Pantoprazole Sodium Enteric-Coated Capsules and Its Related Substances,and study the stability .Methods The chromatographic separation was performed on AgilentC(18)(250 nm×4.6 nm,5 μm),Sodium dihydrogen phosphate buffer(dissolved 1.20 g sodium dihydrogen phosphate in 1 000 mL water,and ajust pH 7.0 with phosphoric acid)—acetonitrile=68∶32,the flow rate was 1.0 mL·min(-1),the detection wavelength was 288 nm,and detective the related substances and the content . Theacceleration test was carried outat temperature (40±2)℃,relative humidity of (75±5)%,and test the result.ResultsThe linear of Pantoprazole Sodium of atorvastatin calcium was (1.0~10.0)×10(-3) g·L(-1),the limits of detection was 0.20 ng,the limit of quantitation was 0.60 ng,the average recovery was 100.57%(RSD=0.41%),Determination of the sample average content was 100.67%,and the related substance was less than 0.2%. The result of stability test that the content was 99.70%~101.16%,the related substance was less than 0.2%.ConeclusionThe method was quick,simple,stable,good repeatability,and it could be used as the content of pantoprazole sodium enteric-coated capsules and its related substances. The stability results conform to the requirement.
Key words:pantoprazole sodium enteric-coated capsules;HPLC;content;related substances;stability
(收稿日期:2015-10-16,修回日期:2015-12-21)
doi:10.3969/j.issn.1009-6469.2016.04.013
猜你喜欢
氯碱工业(2021年6期)2021-12-25
数学物理学报(2021年5期)2021-11-19
石油沥青(2021年4期)2021-10-14
天津医科大学学报(2021年1期)2021-01-26
世界科学技术-中医药现代化(2020年2期)2020-07-25
厦门理工学院学报(2016年1期)2016-12-01
天然产物研究与开发(2016年6期)2016-06-05
现代防御技术(2016年1期)2016-06-01
中国资源综合利用(2016年10期)2016-01-22
现代防御技术(2014年5期)2014-02-28
