
缬沙坦氨氯地平片有关物质研究
2016-05-23 02:54周志勇黄鹤飞
安徽医药 2016年4期
关键词:高效液相色谱
周志勇,黄鹤飞,张 烨
(1.三峡大学医学院药学系;2.三峡大学第一临床医学院药剂科,湖北 宜昌 443003)
◇药物分析◇
缬沙坦氨氯地平片有关物质研究
周志勇1,黄鹤飞2,张烨2
(1.三峡大学医学院药学系;2.三峡大学第一临床医学院药剂科,湖北 宜昌443003)
摘要:目的建立缬沙坦氨氯地平片有关物质含量的测定方法。方法采用高效液相色谱法,Agilent1220高效液相,(Eclipse plus,250 mm×4.6 mm,5 μm)色谱柱,以磷酸盐缓冲液(取三乙胺10 mL加水至1 000 mL,用磷酸调节pH值至2.8)为流动相A,以甲醇—乙腈(70∶30)为流动相B,进行线性梯度洗脱,流速为1.0 mL·min(-1),检测波长为237 nm。结果氨氯地平与杂质D分离度为14.44,缬沙坦与杂质B的分离度为9.46,且各物质之间有较好的分离;空白辅料对有关物质测定无干扰,专属性良好;强制降解试验中,杂质可与主峰完全分离。结论该方法结果准确,重复性好,可靠性强,可用于测定缬沙坦氨氯地平片中的有关物质。
关键词:缬沙坦氨氯地平;有关物质;高效液相色谱
缬沙坦氨氯地平片是由诺华公司研发,并于2006年12月获得美国FDA批准用于治疗原发性高血压,用于单药治疗不能充分控制血压的患者。由于该药疗效确切,国内开展了大量的该复方制剂仿制药的研发工作[1-3],而有关物质的完整研究资料是申报该复方制剂的重要组成部分,建立有关物质的质量标准以控制有关物质含量。参照缬沙坦氨氯地平片进口标准“有关物质”项下检测的杂质及购得的杂质对照品,将缬沙坦杂质B(结构式见图1)和氨氯地平杂质D(结构式见图2)列入标准考察。本试验采用高效液相色谱法梯度洗脱方式,建立了快速、准确、分离度高的含量测定方法,从而为完善缬沙坦氨氯地平片的质量标准提供了依据。

图1 缬沙坦杂质B结构示意图
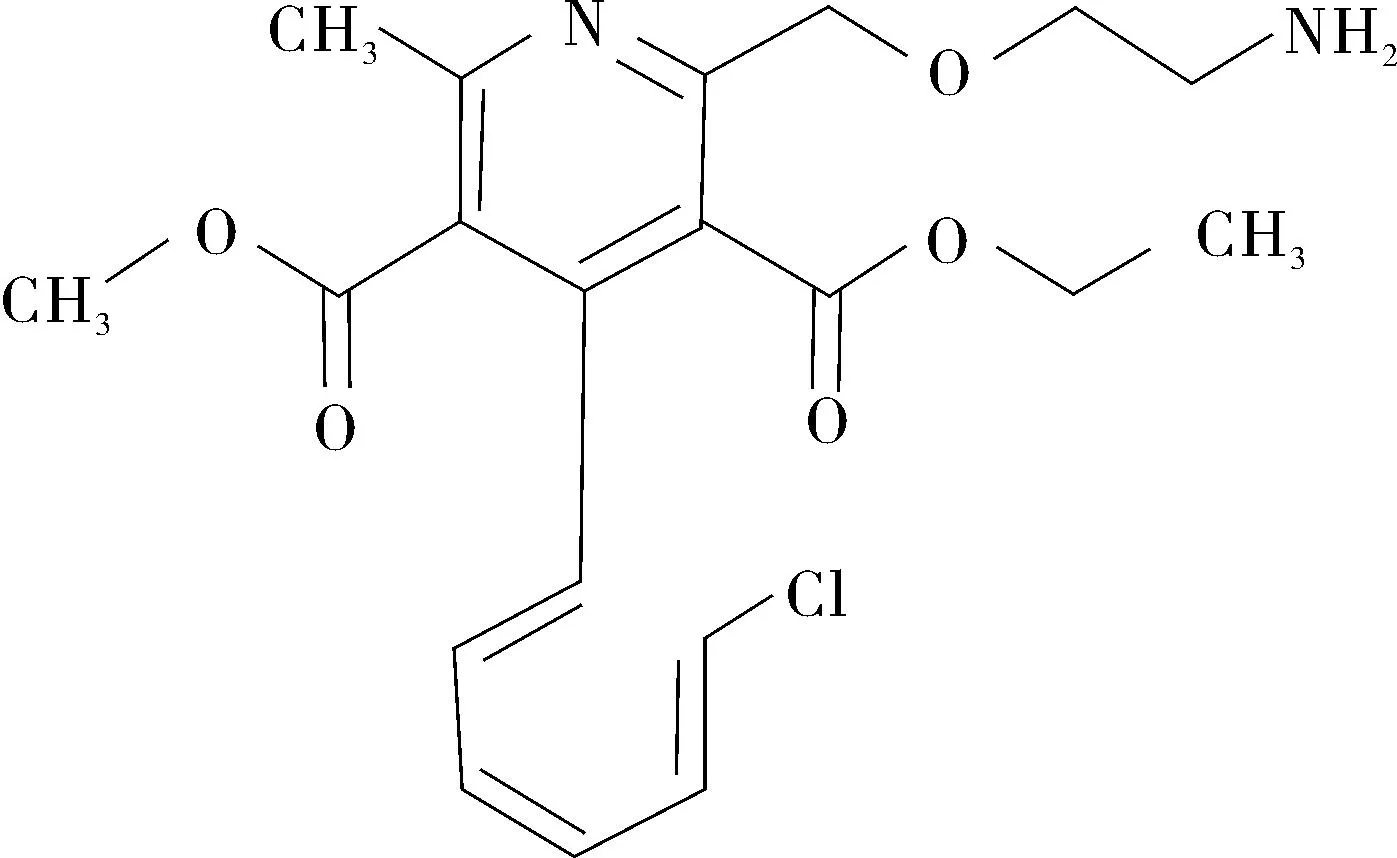
图2 氨氯地平杂质D结构示意图
1仪器与试药
Agilent1220液相色谱仪(安捷伦科技有限公司);FA2004N电子分析天平(上海精密科学仪器有限公司);KQ-500DE超声仪(昆山市超声仪器有限公司);苯磺酸氨氯地平(中国药品生物制品检定所,批号:100374-200903);缬沙坦(中国药品生物制品检定所,批号:100651-200902);氨氯地平杂质D(Kony Pharma,批号:20090901);缬沙坦异构体(Kony Pharma,批号:20080801);乙腈为色谱纯; 水为二次去离子水;其余试剂为分析纯。
2方法与结果[4-10]
2.1色谱条件与系统适用性试验采用Agilent,Eclipse plus色谱柱(250 mm×4.6 mm,5 μm);以磷酸盐缓冲液(取三乙胺10 mL加水至1 000 mL,用磷酸调节pH值至2.8)为流动相A,以甲醇—乙腈(70∶30)为流动相B;检测波长为237 nm;柱温为30℃;进样量10 μL;流速为1.0 mL·min-1,按表1进行线性梯度洗脱。

表1 线性梯度洗脱
精密量取氨氯地平杂质D对照品溶液和缬沙坦杂质B对照品溶液各1 mL置10 mL量瓶中,加缬沙坦氨氯地平对照品溶液溶解并稀释至刻度,摇匀,作为系统适用性溶液。精密量取系统适应性溶液10 μL,注入液相色谱仪,记录色谱图,结果见表2。结果显示,氨氯地平及杂质D和缬沙坦及杂质B的理论塔板数均大于10 000,氨氯地平与杂质D的分离度及缬沙坦与杂质B的分离度均>1.5。
2.2溶液的制备
2.2.1供试品溶液取本品10片,精密称定,研细,精密称取适量(约相当于缬沙坦80 mg,氨氯地平5 mg),置50 mL量瓶中,加流动相A—流动相B(1∶1)适量,超声使溶解,放冷,加流动相A—流动相B(1∶1)稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液。

表2 有关物质系统适用性试验结果
注:分离度1为氨氯地平与杂质D的分离度,分离度2为缬沙坦与杂质B的分离度。
2.2.2对照溶液精密量取供试品溶液1 mL置100 mL量瓶中,加流动相A-流动相B(1∶1)稀释至刻度,摇匀,作为对照溶液。
2.2.3氨氯地平杂质D对照品溶液取氨氯地平杂质D对照品适量,精密称定,加流动相A-流动相B(1∶1)溶解并稀释制成每1 mL中含有氨氯地平杂质D 0.8 mg的对照品溶液。
2.2.4缬沙坦杂质B对照品溶液取缬沙坦杂质B对照品适量,精密称定,加流动相A-流动相B(1∶1)溶解并稀释制成每1 mL中含有缬沙坦杂质B 0.8 mg的对照品溶液。
2.3杂质归属
2.3.1氨氯地平杂质D对照品溶液取氨氯地平杂质D对照品适量,精密称定,加流动相A—流动相B(1∶1)溶解并稀释制成每1 mL中含有氨氯地平杂质D 0.8 mg的对照品溶液。
2.3.2缬沙坦杂质B对照品溶液取缬沙坦杂质B对照品适量,精密称定,加流动相A—流动相B(1∶1)溶解并稀释制成每1 mL中含有缬沙坦杂质B 0.8 mg的对照品溶液。
2.3.3苯磺酸溶液精密量取液体苯磺酸1 mL,加流动相A—流动相B(1∶1)溶解并稀释至100 mL。
2.3.4氨氯地平原料溶液精密称取苯磺酸氨氯地平原料14 mg,置100 mL量瓶中,加甲醇5 mL使溶解,加流动相A—流动相B(1∶1)稀释至刻度,摇匀,作为氨氯地平原料溶液。
2.3.5缬沙坦原料溶液精密称取缬沙坦原料16 mg,置100 mL量瓶中,加甲醇5 mL使溶解,加流动相A—流动相B(1∶1)稀释至刻度,摇匀,作为缬沙坦原料溶液。分别精密量取上述溶液各10 μL,注入液相色谱仪,记录色谱图,结果见表3。苯磺酸的保留时间为2.092 min,氨氯地平杂质D保留时间为7.231 min,缬沙坦杂质B保留时间为13.937 min,未知杂质分别为8.876、9.097、14.817 min。
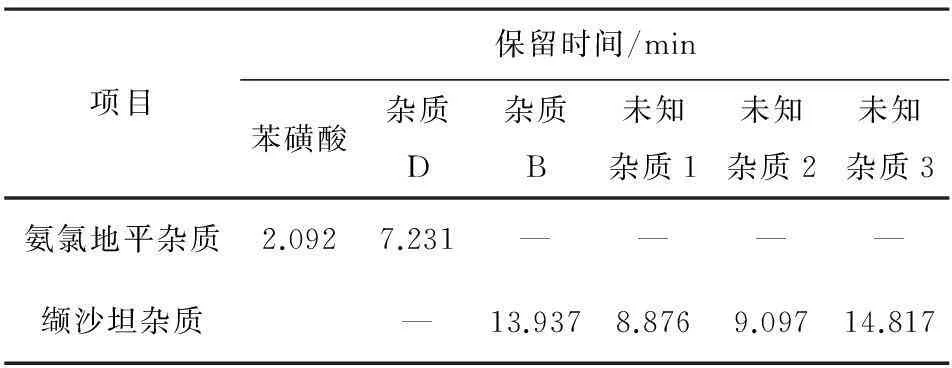
表3 杂质归属结果
注:“—”为未检出。
2.4系统适用性试验
2.4.1缬沙坦氨氯地平对照品溶液精密称取苯磺酸氨氯地平对照品14 mg和缬沙坦对照品16 mg,置100 mL量瓶中,加甲醇5 mL使溶解,加流动相A—流动相B(1∶1)稀释至刻度,摇匀,作为缬沙坦氨氯地平对照品溶液。
2.4.2系统适用性溶液精密量取氨氯地平杂质D对照品溶液和缬沙坦杂质B对照品溶液各1 mL置10 mL量瓶中,加缬沙坦氨氯地平对照品溶液溶解并稀释至刻度,摇匀,作为系统适用性溶液。精密量取系统适应性溶液10 μL,注入液相色谱仪,记录色谱图,结果见表4。
2.5杂质相对保留时间及校正因子因有关物质测定为梯度洗脱,流动相相对固定,未考察不同流动相比例下各杂质的对保留时间,重点考察两个厂家的色谱柱测定杂质的保留时间,并计算校正因子。
色谱柱1:Lichrospher C18(150 mm×64.6 mm,5 μm);色谱柱2:Agilent C18(150 mm×64.6 mm, 5 μm)。
精密量取系统适用性溶液10 μL,注入液相色谱仪,记录色谱图,结果见表5。

表4 有关物质系统适用性试验结果
注:分离度1为氨氯地平与杂质D的分离度,分离度2为缬沙坦与杂质B的分离度。

表5 杂质保留时间考察试验结果
注:RRT(杂质相对保留时间)均为相对于氨氯地平峰的保留时间。
由上述试验结果可知,杂质D相对于氨氯地平的保留时间为0.570~0.647 min,平均值为0.61 min,杂质B相对于氨氯地平的保留时间为1.233~1.325 min,平均值为1.28 min,因此拟定杂质D相对于氨氯地平的保留时间为0.61 min,杂质B相对于氨氯地平的保留时间为1.28 min。
试验测得杂质D的校正因子为2.03,杂质B的校正因子为1.01~1.02,参考缬沙坦氨氯地平片进口标准,确定杂质D的校正因子为2.0,杂质B及其他杂质的校正因子为1.0。
2.6空白辅料干扰试验取空白辅料样品0.083 0 g,置50 mL量瓶中,加流动相A—流动相B(1∶1)溶解并稀释至刻度,作为空白辅料溶液,精密量取10 μL,注入液相色谱仪,记录色谱图,结果可知,空白辅料溶液不出峰,不干扰本品有关物质的测定。
2.7耐用性试验为了考察色谱条件微小的变动能否通过设计的系统适用性试验,确保方法的有效性,故采用改变下列条件考察本方法的耐用性。结果见表6,7。
原色谱条件:用十八烷基硅烷键合硅胶为填充剂,Licrospher C18(150 mm×4.6 mm,5 μm);流动相:以磷酸盐缓冲液(取三乙胺10 mL加水至1 000 mL,用磷酸调节pH值至2.8)为流动相A,以甲醇—乙腈(70∶30)为流动相B,进行梯度洗脱;检测波长:237 nm;流速:1.0 mL·min-1;柱温:30℃;进样量:10 μL。

表6 氨氯地平耐用性试验结果
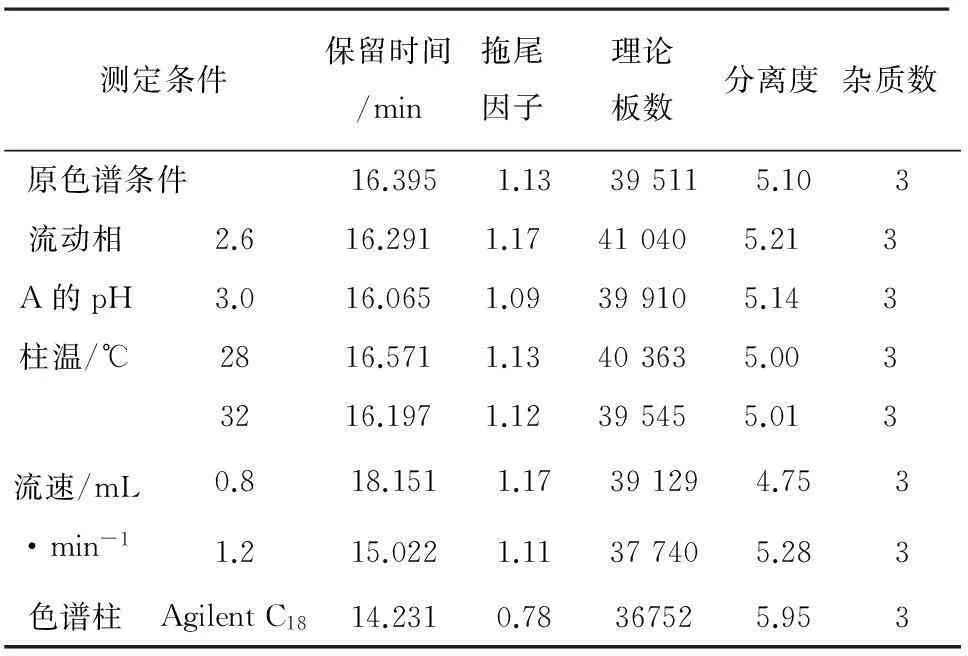
表7 缬沙坦耐用性试验结果
结论:流动相pH、流速及柱温的变化对氨氯地平和缬沙坦的有关物质检查无影响,杂质峰的数量没有增加。表明本方法系统耐用性良好,故确定色谱条件为:流动相A∶磷酸盐缓冲液(取三乙胺10 mL加水至1 000 mL,用磷酸调节pH至2.8),流动相B:甲醇—乙腈(70∶30),梯度洗脱,流速为1.0 mL·min-1;柱温30℃。
2.8检测限取本品供试品溶液,稀释进样至信噪比为3∶1,作为检测限,氨氯地平检测限为0.695ng,缬沙坦检测限为0.775 ng,能满足有关物质检查的要求。
2.9溶液放置稳定性试验取供试品溶液,分别在0、2、4、6、8、10、12 h,精密量取10 μL注入液相色谱仪,记录色谱图,结果显示供试品溶液中氨氯地平和缬沙坦在12 h内基本稳定,RSD=1.36%。
2.10强制降解试验
2.10.1未破坏取本品细粉适量(约相当于缬沙坦80 mg,氨氯地平5 mg),置50 mL带刻度的具塞试管中,加入流动相A—流动相B(1∶1)适量,超声15 min,放冷,加流动相A—流动相B(1∶1)稀释至50 mL,滤过,取续滤液作为破坏前溶液。
2.10.2酸破坏试验取本品细粉适量(约相当于缬沙坦80 mg,氨氯地平5 mg),置50 mL带刻度的具塞试管中,加1 mol·L-1盐酸溶液3 mL,置80℃水浴中加热约30 min,放冷至室温,用1 mol·L-1氢氧化钠溶液调至中性,加流动相A—流动相B(1∶1)稀释至50 mL,滤过,取续滤液作为酸破坏溶液。
2.10.3碱破坏试验操作步骤同酸破坏试验,仅将盐酸换位氢氧化钠,得碱破坏溶液。
2.10.4氧化破坏试验取本品细粉适量(约相当于缬沙坦80 mg,氨氯地平5 mg),置50 mL带刻度的具塞试管中,加30%的双氧水溶液5 mL,静置2 h,加流动相A—流动相B(1∶1)稀释至50 mL,滤过,取续滤液作为氧化破坏溶液。
2.10.5高温破坏试验取本品细粉适量(约相当于缬沙坦80 mg,氨氯地平5 mg),置50 mL带刻度的具塞试管中,加流动相A—流动相B(1∶1)5 mL,置100℃水浴中加热约2 h,放冷至室温,加流动相A—流动相B(1∶1)稀释至50 mL,滤过,取续滤液作为高温破坏溶液。
2.10.6光照破坏试验取本品细粉适量(约相当于缬沙坦80 mg,氨氯地平5 mg),置50 mL带刻度的具塞试管中,置照度为(4 500±500)Lx的光照箱中48 h,加入流动相A—流动相B(1∶1)适量,超声15 min,放冷,加流动相A—流动相B(1∶1)稀释至50 mL,滤过,取续滤液作为光照破坏溶液。
另取空白辅料、缬沙坦和氨氯地平的混合原料(按处方比例混合)、被仿制品细粉,同法操作。分别精密量取上述溶液各10 μL,注入液相色谱仪。

图3 强制降解试验色谱图
图3可见本品在酸、碱、高温、氧化、光照条件下均不稳定,原料带入杂质B,酸破坏降解产物主要有杂质D和相对氨氯地平峰保留时间约为0.24的未知杂质,碱破坏降解产物主要有杂质B、D和相对氨氯地平峰保留时间在0.35~0.42 min之间的未知杂质,氧化破坏降解产物主要有杂质D,高温破坏降解产物主要有杂质B、D和相对氨氯地平峰保留时间约为0.24 min的未知杂质,光照破坏降解产物主要有杂质B、D和一些未知杂质。自制样品降解产物个数和变化趋势与上市样品基本一致。
3讨论
《英国药典》(2009年版)中缬沙坦氨氯地平片有关物质中,缬沙坦与氨氯地平分别记录了4种有关物质,查阅相关文献[11-13],均将氨氯地平杂质D和缬沙坦杂质B列入考察标准中。
本试验分别从系统适应性、专属性以及强制降解试验方面分别考察了HPLC测定缬沙坦氨氯地平片有关物质的方法,结果显示的目标色谱峰与有关物质分离效果良好,本HPLC法测定有关物质方法简便、快捷、可靠,可用于缬沙坦氨氯地平复方片质量标准中有关物质的检测。
参考文献:
[1]吕晴,郝玲花,李东辉,等.高效液相色谱法测定缬沙坦氢氯噻嗪片有关物质[J].中国新药杂志,2014,23(15):1754-1757.
[2]魏春燕,沈利,王延松.HPLC法测定复方氨氯地平缬沙坦片有关物质[J].中国药师,2011,14(8):1142-1144.
[3]王燕,毛白杨,狄斌,等. LC-MS/MS法分析复方缬沙坦氨氯地平片中的有关物质[J].新疆医科大学学报,2013,36(5):611-617.
[4]国家药典委员会.中国药典(二部)[S].北京:中国医药科技出版社,2010:1136-1137.
[5]张少桦,王锦刚,蒋海松.HPLC 法检查左乙拉西坦缓释片的有关物质[J].中国药房,2014,25(13):1212-1215.
[6]邓紫薇,卢欣,李美珍,等.复方氢氯噻嗪片剂的质量控制[J].天津医科大学学报,2012,18(4):514-517.
[7]荆小燕,王勇军,徐丽洁,等.高效液相色谱法测定缬沙坦氨氯地平片中的有关物质[J].中国新药,2013,22(19):2323-2327.
[8]刘宪勇,刘世军,孙克明,等. HPLC法测定奥拉西坦原料药中的有关物质[J].中国药房,2014,25(1):66-68.
[9]彭熙琳,吴品江,周唯兰,等.应用高效液相色谱法测定缬沙坦氨氯地平片有关物质[J].重庆医学,2014,43(17):2571-2577.
[10] 付长华,杨琴香,蔡华吉.HPlC法测定苯磺酸左旋氨氯地平片的含量及有关物质[J].江西中医学院学报,2013,25(6):58-60.
[11] 张翠兰.高效液相色谱法测定那格列奈胶囊的有关物质[J].安徽医药,2015,19(12):2310-2313.
[12] 唐杰,邱幸琛.苯磺酸氨氯地平片有关物质测定[J].海峡药学,2012,24(10):96-98.
[13] 张辉,丁双婷.HPLC梯度洗脱法检查缬沙坦氢氯噻嗪分散片的有关物质[J].食品与药品,2012,14(9):343-345.
Research on related substance of valsartan Amlodipine
ZHOU Zhi-yong1,HUANG He-fei2,ZHANG Ye2
(1.CollegeofMedicalScience,ChinaThreeGorgesUniversity; 2.DepartmentofPharmacy,SecondCollegeofClinicalMedicalScience,ChinaThreeGorgesUniversity,Yichang,Hubei443003,China)
Abstract:Objective Aimed to establish an analytical method for related substances of valsartan Amlodipine tablets. Methods The sample was determined by HPLC (Agilent1220). The analysis was performed on Eclipse plus C(18 )column (250 mm×4.6 mm, 5 μm) with the detection wavelength at 237 nm. Using phosphate buffer (10 ml triethylamine volumetric to 1 000 mL,adjusted to pH 2.8 with phosphoric acid) as mobile phase A, methanol - acetonitrile (70∶30) as mobile phase B, and linear gradient, mobile phase was initiated at a flow rate of 1.0 mL·min(-1).Results The resolution of amlodipine and impuritie D was 14.44;the resolution of amlodipine and impuritie B was 9.46, and there was good resolution between each substance. Conclusions This method is accurate, reproducible and reliable, which can be used to determine the related substances of valsartan Amlodipine tablets.
Key words:valsartan amlodipine;related substance;HPLC
(收稿日期:2015-10-20,修回日期:2016-02-26)
doi:10.3969/j.issn.1009-6469.2016.04.010
猜你喜欢
分析化学(2017年1期)2017-02-06
中国医药导报(2016年30期)2016-12-28
热带农业科学(2016年10期)2016-12-12
分析化学(2016年7期)2016-12-08
上海医药(2016年21期)2016-11-21
科学与财富(2016年28期)2016-10-14
上海医药(2016年3期)2016-03-23
现代仪器与医疗(2015年4期)2015-07-15
