
基于SMRT测序技术的16S rRNA基因全长测序及其分析方法
2017-08-07 23:22唐勇刘旭
生物技术通报 2017年8期
唐勇刘旭
(1. 乐山职业技术学院,乐山 614000;2. 乐山丰野农业科技有限责任公司,乐山 614000;3. 乐山市农业局,乐山 614000)
基于SMRT测序技术的16S rRNA基因全长测序及其分析方法
唐勇1,2刘旭3
(1. 乐山职业技术学院,乐山 614000;2. 乐山丰野农业科技有限责任公司,乐山 614000;3. 乐山市农业局,乐山 614000)
被称为第三代测序技术的单分子测序是最近几年发展起来的高通量测序技术。其中,由Pacbio BioSciences公司开发的单分子实时测序技术(SMRT)是最先商用的技术。SMRT测序技术通过对模板序列循环测序产生环形一致序列(CCS),成功克服第三代测序技术准确率低的弊病。通过SMRT测序技术,科学家可以更深入准确地探究复杂环境微生物的结构和功能。介绍SMRT测序技术在微生物16S rRNA基因测序中的优势和劣势,并就基于SMRT测序技术所得的全长16S rRNA基因序列的质量控制、错误序列排除、聚类和注释分析等重要分析环节进行概述,同时,提出利用SMRT测序技术研究复杂环境微生物可能存在的问题及其解决方法,期望能为研究人员提供参考。
单分子实时测序技术;PacBio RS Ⅱ;第三代测序技术;环形一致序列
16S rRNA基因是原核生物所特有的基因,并且在原核生物中具有极高的拷贝数[1]。全长1 542 nt的DNA序列包含9个间隔的高变区,兼具特异性和保守性的16S rRNA基因序列作为微生物标记被广泛应用于研究中[2]。相比DNA探针、变性梯度凝胶电泳和Sanger测序等方法,高通量测序技术在16S rRNA基因序列研究中体现出极大的优势[3]。以Roche[4],Illumina[5]等为代表的第二代测序技术将16S rRNA基因测序的通量大幅提高,为研究者提供对特定环境微生物进行全面分析的可能。目前,第二代测序技术已经成为环境微生物研究的主流手段。但是,第二代测序技术在16S rRNA基因测序中存在的缺陷也不可忽视——测序片段短。第二代测序平台中,测序片段最长的是Roche公司开发的454 GS FLX+测序仪,其测序片段长度仅为700 bp。过短的测序片段使得研究人员在微生物16S rRNA基因测序中,只能选择部分高变区进行研究,这对研究结果的准确性有较大影响。
单分子测序技术的出现为解决第二代测序技术中测序读长短的问题提供了可能。单分子测序技术也被称为第三代测序技术(Thrid-generation sequencing,TGS),包括Oxford Nanopore的纳米孔测序技术[6]和单分子实时测序技术(SMRT)[7]等,其中,纳米孔测序技术受限于其测序错误过高的问题,在微生物16S rRNA基因测序等领域应用较少[8]。目前,只有PacBio的RS系列测序平台被大量商用。PacBio RSⅡ测序仪的测序长度可达到20 000 bp,这为基于16S rRNA基因测序的微生物研究提供更好的选择。本文将就PacBio的SMRT测序技术在16S rRNA基因测序中的优势综述,然后介绍PacBio RS系列测序仪测得的全长16S rRNA序列分析方法及其应用,最后提出目前存在的问题和可能的解决方案,以期为研究人员采用SMRT测序技术研究微生物16S rRNA基因提供参考。
1 SMRT测序技术在16S rRNA基因测序中的优势
1.1 读长覆盖全长16S rRNA基因
基于16S rRNA基因的微生物多样性的研究中,测序部分高变区是目前最常用的方法。研究表明不同的高变区(V1-V9)对不同分类下的微生物分辨率不同[9]。研究表明,V1-V3区域所包含的信息量最接近全长16S rRNA基因的信息量,但是,两者依然存在较大的差别[10]。因此,测序全长16S rRNA基因序列是最理想的方法。基于SMRT测序技术的PacBio RSⅡ测序仪在使用P6/C4试剂的情况下平均reads长度超过10 kb,N50(序列长度中位数)超过20 kb,最长达到60 kb[11,12]。因此,该测序技术可以轻易完成16S rRNA基因全长序列的测序。Mosher等[13]使用PacBio RSⅡ及P4/C2试剂测得沉淀物样本微生物16S rRNA基因片段平均长度为1 419-1 431 bp。在Benitez-Paez等和Schloss等[14,15]的研究中,使用全长16S rRNA基因序列对物种多样性、微生物组成和微生物进化开展研究,对实验的准确性和分辨率能够带来显著的提升。同时,将研究深入到种水平,而不是局限在属水平上[16]。
1.2 相对可靠度的16S rRNA基因序列
第三代测序技术最初无法运用于16S rRNA基因序列测序的原因是高测序错误率。基于纳米孔测序技术的MinION测序仪reads错误率高达40%[17],PacBio的reads测序错误达到15%,单碱基错误率1%[18]。显然,如此高的错误率无法满足16S rRNA基因用于微生物种属的鉴定。
SMRT测序技术中的测序错误以单碱基的插入和缺失为主[18],而且呈现随机分布的模型[19]。因此,采用循环测序的方法对同一条16S rRNA基因模板多次测序,再通过多重比对方法获取环形一致序列(Circular consensus sequence,CCS),这样可以大幅减少测序引起的碱基错误。据Eid等[20]2009年报道,碱基测序深度达到15X即可有效提高正确率至99.3%。Schloss等[15]在分析中进一步采用质控、过滤和预聚类等生物信息分析步骤,将碱基错误率降低至0.03%。尽管,此方法的碱基错误率仍然高于Miseq、454等第二代测序平台,但是其错误率已经在可接受的范围之内。
1.3 测序速度更快
SMRT测序技术测序速度快,可以达到10 bp/s,能够缩短测序工作的周期。相比第二代测序技术数天的测序时间,SMRT测序技术每个run的测序时间仅为0.5-4 h之间[11,21]。虽然科研中大多数情况对测序时间不敏感,但是对于临床上需要快速对微生物进行鉴定诊断的情况下,SMRT测序技术更具备应用的优势。
2 基于SMRT的16S rRNA基因测序数据分析流程
2.1 质量控制
最新的PacBio RSⅡ测序仪的下机数据为bam格式(老版本软件的输出格式为h5,可以使用bax2bam将h5格式数据文件转换为BAM格式)。PBCSS(https://github.com/PacificBiosciences/unanimity)是PacBio公司开发的PacBio raw reads分析工具,用于获取CSS reads并同时进行质量控制的软件。该软件以SAM格式为输入和输出文件,可以完成预测质量值过滤、序列长度过滤、测序深度(PASS)过滤、CSS reads预测准确度过滤以及识别正负链。除此之外,还可以结合NGS QC Toolkit[22]、FASTQC[23]等软件进行质量控制。
通常CCS reads质量可以从以下几个方面控制:(1)测序所得的16S rRNA CCS reads的长度应该为全长或者接近全长,远远低于或者超过预期长度的片段应该过滤;(2)CCS reads预测准确度大于99%[13,24];(3)CCS的测序深度(PASS)至少为3,在测序数据充足的情况下,推荐值为10[15];(4)碱基质量至范围在2-93之间,考虑碱基错误为随机分布,因此,通常采用CCS reads所有碱基平均质量值作为过滤条件,推荐值为30;(5)包含模糊碱基(N)的序列同样考虑进行过滤。
2.2 鉴定并过滤嵌合体
16S rRNA基因序列扩增和SMRT测序过程中,依然不可避免地会产生嵌合体序列(Chimera),嵌合体过滤仍然是不可缺少的分析步骤。考虑16S rRNA基因序列测序片段显著增加,嵌合体序列的识别率也将得到提升。UCHIME[25]仍然是嵌合体检测使用最普遍的软件[26],结合SILVA[27]、RDP[28]和greengenes[29]等数据库可以完成有参的嵌合体检测分析。同时,考虑数据库完整性问题,也可以使用UCHIME基于reads丰度的de novo检测方法识别嵌合体序列。
2.3 CCS reads预聚类与低丰度序列过滤
预聚类的目的是通过将遗传距离极小的CCS reads聚类在一起,以避免CCS reads中少量的碱基错误对后续的分析造成影响。在微生物16S rRNA基因序列聚类中,将相似度大于97%的序列划分为同一种,来自同一种的16S rRNA基因序列差异可能由种内遗传变异或者测序错误引起。因此,选择更高相似度作为预聚类阈值可以将这部分序列差异过滤掉[30]。在SMRT技术测序中,经过序列矫正之后,序列的正确率可以达到99.3%[20]。因此,理论上选择99%的相似度作为预聚类阈值可以排除错误碱基的影响。
低丰度序列往往更倾向于来自人工序列[31],当然,也不否认在低丰度序列中包含少数来自于稀有微生物的16S rRNA序列[32]。但是从环境微生物的研究角度考虑,过滤低丰度序列的利大于弊,而且这一方案在基于第二代测序技术的研究中取得较好的结果[33,34]。基于SMRT测序技术的CCS reads的错误序列随机性更强,因此,可以推测随机错误引起的错误序列更倾向于表现为低丰度。虽然,目前没有实验报道这一假设,但是为了获得更为保守的微生物多样性结果,使用过滤低丰度CCS reads的策略更为妥当[13]。
对CCS reads预聚类和低丰度序列过滤能够大幅度减少错误序列对OTUs聚类的干扰。但是,应该谨慎选择预聚类阈值和过滤的丰度值,避免造成微生物多样性结果低估。以上两个步骤能够在UPARSE[35]、MOTHUR[36]等软件中完成。
2.4 OTUs聚类与注释
获得预处理的CCS reads之后,按97%相似度进行OTUs聚类分析,可以选择MOTHUR[36]、UPARSE[35]或者QIMME等任意软件完成。其中,选择OTUs代表序列有两种方法可选,分别是使用高丰度序列和OTUs内一致性序列[15]。目前,在已有的基于SMRT测序技术的16S rRNA基因序列研究中,两种方法都有采用。
对全长16S rRNA基因进行注释时,基于朴素贝叶斯分类器的RDP-Classifier[37]依然是最有效的工具[38]。而在数据库的选择上,RDP是更新最快,使用最广泛的软件,在属及以上水平注释的准确性最高。而greengene虽然更新速度慢,而且包含的参考序列少,但是却有11%的序列具有种水平的注释信息,这是其他数据库无法比拟的优势[15]。此外,grengene的16S rRNA序列选自NCBI数据库中长度大于1 200 bp的序列,长度更接近全长序列;相比之下,RDP只有不超过44%的细菌和15.3%的真菌16S rRNA基因序列长度超过1 200 bp[10]。因此,考虑测序长度为全长序列,参考序列应该选择更长的16S rRNA基因序列,或者综合多个数据库进注释(表1)。
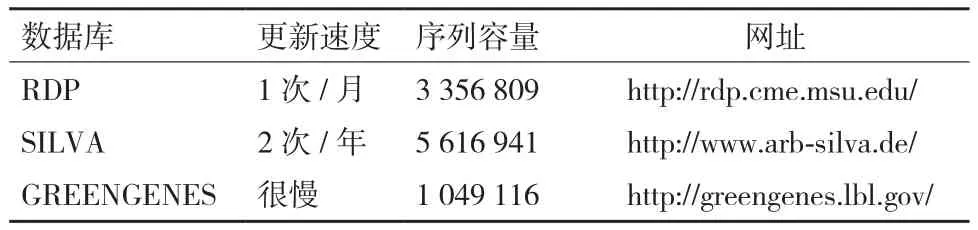
表1 主要16S rRNA数据库
3 SMRT测序技术存在的问题
3.1 测序错误
SMRT测序技术的优势在于同时兼顾测序长度长和测序错误相对较低的优点[39]。但是,单从测序错误率方面讨论,其测序错误问题还需要进一步改善。基于SMRT测序技术中的碱基错误为随机分布的假设,通过提高循环测序深度(PASS)可以减少碱基错误。但是,Schloss等[15]的实验发现,SMRT测序技术也可能存在系统错误,从而导致错误碱基无法通过提高循环测序深度排除这部分碱基错误。除此之外,嵌合体序列也是不可避免的问题,目前暂时没有办法完全排除嵌合体的干扰。
3.2 数据库完整性
目前,16S rRNA基因注释数据库的完整性普遍较差,而且数据库中的参考序列长度较短。这直接造成两个问题:(1)OTUs的注释率偏低,而能够注释到种水平上的序列更少;(2)尽管可以获得全长的16S rRNA基因序列,但参考序列长度不足导致注释的准确性大打折扣。随着SMRT测序技术的发展,越来越多的全长16S rRNA基因序列被准确测序,这也许能够为提高16S rRNA基因数据库的完整性提供新的契机。
3.3 测序成本偏高
PacBio SR Ⅱ测序平台单个cell的测序价格低,但是单个cell的数据输出量少,因此,单个碱基的价格要远高于Illumina等第二代测序平台[11]。满足研究需求的测序量所付出的测序成本依然偏高,这是阻碍SMRT测序技术在16S rRNA基因测序中推广应用的主要因素。随着PacBio SR测序平台的升级,测序量和测序质量不断提升,测序成本逐渐降低,相信基于SMRT测序技术的全长16S rRNA基因测序会越来越多地运用于临床和研究当中。
4 展望
从2005年,454推出第一台商用高通量测序仪开始,测序技术飞速发展,目前sanger测序技术、第二代测序技术和第三代测序技术凭借各自优势在研究中扮演着不同的角色。基于16S rRNA基因测序的微生物研究中,第二代测序技术仍然是主力。以SMRT技术为代表的第三代测序技术兼具测序长度和测序通量优势,为16S rRNA基因全长测序打开一扇新的窗户。基于SMRT测序技术的微生物16S rRNA基因测序可以有效提高环境微生物研究的分辨率[15],将更多微生物注释到种水平,并且提高物种丰度预测的准确性。基于此,结合微生物物种参考基因组,有望直接使用16S rRNA测序替代环境微生物宏基因组测序,即直接使用16S rRNA基因数据研究微生物基因水平和代谢通路水平差异[40]。
目前,SMRT测序技术自身仍然具有改进的空间,例如:减少测序错误、增加每个cell测序量等。而对于测序之后的进一步分析,也存在较多问题需要解决。首先,针对PacBio测序所得的CCS reads没有专门的分析软件。虽然,绝大部分分析环节可以使用mothur[36]等软件处理,但是,其中分析环节的细节还需要进一步研究。例如,由于全长16S rRNA基因序列之间存在大量非高边区,使用97%作为OTU聚类分析阈值可能导致物种数量被低估。其次,数据库完整性不足可能导致全长16S rRNA基因序列效果大打折扣,其中包括两个方面:其一,数据库注释物种总量不,导致可注释物种减少;其二,数据库内的参考序列长度不足,引起序列注释偏差。
[1]Klappenbach JA, Saxman PR, Cole JR, et al. rrndb:the ribosomal RNA operon copy number database[J]. Nucleic Acids Research, 2001, 29(1):181-184.
[2]Sogin ML, Morrison HG, Huber JA, et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”[J]. Proceedings of the National Academy of Sciences, 2006, 103(32):12115-12120.
[3]Roh SW, Abell GC, Kim K-H, et al. Comparing microarrays and next-generation sequencing technologies for microbial ecology research[J]. Trends in Biotechnology, 2010, 28(6):291-299.
[4]Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors[J]. Nature, 2005, 437(7057):376-380.
[5]Bentley DR. Whole-genome re-sequencing[J]. Current Opinion in Genetics & Development, 2006, 16(6):545-552.
[6]Clarke J, Wu H-C, Jayasinghe L, et al. Continuous base identification for single-molecule nanopore DNA sequencing[J]. Nature Nanotechnology, 2009, 4(4):265-270.
[7]McCarthy A. Third generation DNA sequencing:pacific biosciences’ single molecule real time technology[J]. Chemistry & Biology, 2010, 17(7):675-676.
[8]Mikheyev AS, Tin MM. A first look at the Oxford Nanopore MinION sequencer[J]. Molecular Ecology Resources, 2014, 14(6):1097-1102.
[9]Chakravorty S, Helb D, Burday M, et al. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria[J]. Journal of Microbiological Methods, 2007, 69(2):330-339.
[10]Kim M, Morrison M, Yu Z. Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes[J]. J Microbiol Methods, 2011, 84(1):81-87.
[11]Rhoads A, Au KF. PacBio sequencing and its applications[J]. Genomics, Proteomics & Bioinformatics, 2015, 13(5):278-289.
[12]Roberts RJ, Carneiro MO, Schatz MC. The advantages of SMRT sequencing[J]. Genome Biology, 2013, 14(7):405.
[13]Mosher JJ, Bowman B, Bernberg EL, et al. Improved performance of the PacBio SMRT technology for 16S rDNA sequencing[J]. Journal of Microbiological Methods, 2014, 104:59-60.
[14]Benitez-Paez A, Portune KJ, Sanz Y. Species-level resolution of 16S rRNA gene amplicons sequenced through the MinIONTMportable nanopore sequencer[J]. Gigascience, 2016, 5(1):1-9.
[15]Schloss PD, Jenior ML, Koumpouras CC, et al. Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system[J]. PeerJ, 2016, 4:e1869.
[16]Lee CH, Bowman B, Hall R, et al. Developments in PacBio® metagenome sequencing:Shotgun whole genomes and full-length 16S[C]. International Plant and Animal Genome Conference Asia, 2014.
[17]Laver T, Harrison J, O’Neill P, et al. Assessing the performance of the Oxford Nanopore Technologies MinION[J]. Biomolecular Detection and Quantification, 2015, 3:1-8.
[18]Koren S, Schatz MC, Walenz BP, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads[J]. Nature Biotechnology, 2012, 30(7):693-700.
[19]Ross MG, Russ C, Costello M, et al. Characterizing and measuring bias in sequence data[J]. Genome Biology, 2013, 14(5):1.
[20]Eid J, Fehr A, Gray J, et al. Real-time DNA sequencing from single polymerase molecules[J]. Science, 2009, 323(5910):133-138.
[21]Quail MA, Smith M, Coupland P, et al. A tale of three next generation sequencing platforms:comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers[J]. BMC Genomics, 2012, 13(1):341.
[22]Patel RK, Jain M. NGS QC Toolkit:a toolkit for quality control of next generation sequencing data[J]. PLoS One, 2012, 7(2):e30619.
[23]Andrews, S. FastQC:a quality control tool for high throughput sequence data[EB]. http://www.bioinformatics.babraham.ac.uk/ projects/fastqc/.
[24]Bowman B, Shin MY, Lee JE, et al. Analysis of full-length metagenomic 16S genes by SMRT®sequencing[J]. Chemistry, 2013, 4:C2.
[25]Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics, 2011, 27(16):2194-2200.
[26]Haas BJ, Gevers D, Earl AM, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons[J]. Genome Research, 2011, 21(3):494-504.
[27]Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNAgene database project:improved data processing and webbased tools[J]. Nucleic Acids Research, 2013, 41(D1):D590-D596.
[28]Maidak BL, Cole JR, Lilburn TG, et al. The RDP-II(ribosomal database project)[J]. Nucleic Acids Research, 2001, 29(1):173-174.
[29]DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimerachecked 16S rRNA gene database and workbench compatible with ARB[J]. Applied and Environmental Microbiology, 2006, 72(7):5069-5072.
[30]Bowman JS, Rasmussen S, Blom N, et al. Microbial community structure of Arctic multiyear sea ice and surface seawater by 454 sequencing of the 16S RNA gene[J]. The ISME Journal, 2012, 6(1):11-20.
[31]Tedersoo L, Nilsson RH, Abarenkov K, et al. 454 Pyrosequencing and Sanger sequencing of tropical mycorrhizal fungi provide similar results but reveal substantial methodological biases[J]. New Phytologist, 2010, 188(1):291-301.
[32]Lücking R, Lawrey JD, Gillevet PM, et al. Multiple ITS haplotypes in the genome of the lichenized basidiomycete Cora inversa(Hygrophoraceae):fact or artifact?[J]. Journal of Molecular Evolution, 2014, 78(2):148-162.
[33]Unterseher M, Jumpponen A, Öpik M, et al. Species abundance distributions and richness estimations in fungal metagenomicslessons learned from community ecology[J]. Molecular Ecology, 2011, 20(2):275-285.
[34]Kunin V, Engelbrektson A, Ochman H, et al. Wrinkles in the rare biosphere:pyrosequencing errors can lead to artificial inflation of diversity estimates[J]. Environmental Microbiology, 2010, 12(1):118-123.
[35]Edgar RC. UPARSE:highly accurate OTU sequences from microbial amplicon reads[J]. Nature Methods, 2013, 10(10):996-998.
[36]Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur:open-source, platform-independent, community-supported software for describing and comparing microbial communities[J]. Applied and Environmental Microbiology, 2009, 75(23):7537-7541.
[37]Wang Q, Garrity GM, Tiedje JM, et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Applied and Environmental Microbiology, 2007, 73(16):5261-5267.
[38]Liu Z, DeSantis TZ, Andersen GL, et al. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers[J]. Nucleic Acids Research, 2008, 36(18):e120.
[39]Burke CM, Darling AE. A method for high precision sequencing of near full-length 16S rRNA genes on an Illumina MiSeq[J]. Peer J, 2016, 4:e2492.
[40]Langille MG, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences[J]. Nature Biotechnology, 2013, 31(9):814-821.
(责任编辑 李楠)
Full-length Sequencing of 16S rRNA Gene and Its Analysis Based on the SMRT Sequencing Technology
TANG Yong1,2LIU Xu3
(1. Leshan Vocational & Technical College,Leshan 614000;2. Leshan Fengye Agricultural Technology CO.,Ltd,Leshan 614000;3. Agricultural Bureau of Leshan,Leshan 614000)
Single-molecule sequencing,called as third-generation sequencing technology,is a high-throughput technique developed in last few years. Of them,single-molecule real-time(SMRT)sequencing technology,developed by Pacific BioSciences(PacBio),is the first commercial technology. SMRT sequencing technology could successfully overcome the disadvantage of low accuracy in the third-generation sequencing technology,by generating circular consensus sequence(CCS)through cycle sequencing the template sequence. Therefore,SMRT sequencing technology will allow scientists to profoundly and accurately study the structures and functions of microbial communities in complex environment. Here,we introduced the advantages and disadvantages of SMRT sequencing technology in 16S rRNA gene sequence of microorganism,and summarized the important steps,such as quality control,filtering of error tags,clustering analysis,annotation analysis,etc. of full-length 16S rRNA gene sequence acquired by SMRT sequencing technology. In addition,we pointed out the problems and the feasible solutions while applying SMRT sequencing technology in the study of microbial in complex environment,aiming at providing references for researchers in this field.
single-molecule real-time sequencing technology;PacBio RS II;third-generation sequencing technology;circular consensus sequence
10.13560/j.cnki.biotech.bull.1985.2017-0036
2017-01-20
乐山市科技局重点研究项目(16JSC128)
刘旭,女,硕士,研究方向:特种经济动物养殖;E-mail:331424471@qq.com
唐勇,男,博士,研究方向:营养生理;E-mail:ty20042028@163.com
猜你喜欢
传染病信息(2021年6期)2021-02-12
教学考试(高考生物)(2020年6期)2020-11-23
数学小灵通(1-2年级)(2020年9期)2020-10-27
食品与生物技术学报(2020年8期)2020-01-06
科学24小时(2019年5期)2019-06-11
科海故事博览·下旬刊(2019年6期)2019-04-16
发明与创新(2019年9期)2019-03-26
作文大王·低年级(2017年11期)2017-12-05
小学生学习指导(低年级)(2017年12期)2017-11-22
读写算(上)(2015年6期)2015-11-07
