
磷腈型苯并噁嗪的合成及其性能研究
2018-11-16 06:10,
四川化工 2018年5期
,
(1.四川东材科技集团股份有限公司,四川绵阳,621000;2.国家绝缘材料工程技术研究中心,四川绵阳,621000;3.西南科技大学,四川绵阳,621010)
苯并噁嗪树脂具有较高的耐热性、优异的电学性能、低吸水率、开环固化无小分子放出、体积收缩率低等优点[1~3],但普遍存在固化起始温度高,固化后阻燃级别仅V-1级,热稳定性较差等缺点[4~6]。为了弥补苯并噁嗪特性的不足,在苯并噁嗪中加入阻燃剂,可有效提高阻燃等级,但外添加方式会带来相容性问题,导致基体耐热性变差,在长期使用过程中,迁移析出现象明显[7]。近年来不少研究[8~11]致力于将磷腈基团引入苯并噁嗪结构中,开发了不同类型的磷腈型苯并噁嗪,应用检测结果表明,这些磷腈型苯并噁嗪在阻燃性、热稳定性等方面均优于传统苯并噁嗪,已成为苯并噁嗪阻燃研究的热点之一。
本研究在前期合成高支化磷腈苯并噁嗪的工作基础上[11],降低支化度,设计合成出三种噁嗪环数量不同的磷腈型苯并噁嗪,采用红外光谱、核磁氢谱、示差扫描量热分析、热重分析、极限氧指数和垂直燃烧试验对产物进行了表征,探讨了热固化行为,以及固化物结构与热稳定性和阻燃性之间的关系。
1 实验部分
1.1 试剂与仪器
苯酚、对羟基苯甲醚,苯胺、甲醛、乙腈、乙醇、二氯甲烷、甲苯、碳酸钾、三溴化硼,4-甲基-2-苯基咪唑、氢氧化钠等均为分析纯;六氯环三磷腈(HCCP),参照文献[12]自制;双酚A型苯并噁嗪 (Ba),参照文献[2]自制;双酚F型苯并噁嗪 (Bf),参照文献[13]自制。
Nicolet-5700型红外光谱仪 (美国热电仪器公司),KBr压片法;AV600型核磁共振波谱仪 (瑞士Bruker公司),溶剂:CDCl3,内标:TMS;DSC 200型差示量热仪和 TG 209型热重分析仪(德国NETZSCH公司),空气气氛,升温速率10℃/min。
1.2 合成路线(见图1)1.3 合成步骤
D1的合成:将8.7 g HCCP和41.5 g无水碳酸钾加入100 mL乙腈中,在40℃下滴加含9.4 g苯酚的50 mL乙腈溶液,滴毕65℃搅拌5 h,然后加入 8g 对羟基苯甲醚,升温至回流反应5h,过滤,旋干后用二氯甲烷溶解,依次碱洗和水洗,浓缩干燥得淡黄色胶状物16.7 g,产率88.8%。
T1的合成:参照D1的合成,将苯酚用量减少到7 g,对羟基苯甲醚的用量增加到11.8 g,得黄色胶状物17.3 g,产率88.2%。
F1的合成:参照D1的合成,将苯酚用量减少到5.7 g,对羟基苯甲醚的用量增加到12.4g,得棕色胶状物18.3 g,产率90.1%。

图1 磷腈型苯并噁嗪的合成路线
1.3.2 中间体酚羟基化芳氧基磷腈(Ⅱ)的合成
D2的合成:N2保护,冰盐浴冷却下(-10℃ ~0℃),将3.8g D1加入60 mL二氯甲烷中,滴加含1.4mL三溴化硼的60 mL二氯甲烷溶液,滴毕25℃反应3 h,水洗2次,浓缩干燥得淡黄色固体3.5 g,产率96.8%。
T2的合成:参照D2的合成,将三溴化硼的用量增加到1.9 mL,得黄色固体3.5 g,产率94.8%。
F2的合成:参照D2的合成,将三溴化硼的用量增加到2.4mL,得棕色固体3.6 g,产率95.1%。
1.3.3 产物磷腈型苯并噁嗪(Ⅲ)的合成
2015年张兰花从连队住进了团部的楼房,她不仅拥有了农用汽车、小汽车,还在库尔勒市购置了一套100余平米的楼房。“农忙时节种地住团部、节假日住城市,冬闲时节到内地旅游观光,观赏名山大川。”张兰花说,“这些年来,是团场这片热土滋养了我,给了我那么多的惊喜、感动和荣誉,我将一辈子报答团场,以一名党员的模范作用,带动和帮助更多的职工群众一起致富奔小康”。
D3的合成:冰浴冷却下(0℃ ~10℃),在3.3g甲醛和50 mL甲苯的混合液中,滴加1.9g苯胺后搅拌1 h,加入含7.3 g D2的150 mL甲苯溶液,加热至回流反应20 h,依次碱洗和水洗,浓缩干燥后得黄色胶状物8.5 g,产率88.6%。
T3的合成:参照D3的合成,将甲醛用量增加到4.9 g,苯胺用量增加到2.8 g,得橘黄色胶状物9g,产率82.1%。
F3的合成:参照D3的合成,将甲醛用量增加到6.5g,苯胺用量增加到3.7 g,得红棕色固体9.3g,产率75.6%。
1.4 固化物制备
分别取等量的D3、T3、F3、Ba和Bf,放入电热烘箱中进行分段固化,固化温度设定为:130℃/1h,200℃/2h,220℃/1h,240℃/1h,固化后产物依次用PD3、PT3、PF4、PBa和PBf标记。
2 结果与讨论
2.1 产物的结构表征
图2为中间体及产物的红外谱图,其中D1的的红外光谱如图2(a)所示:3004 cm-1是苯环的C-H伸缩振动峰,2960和2830 cm-1是CH3的C-H伸缩振动峰,1593、1506和1463 cm-1是苯环骨架变形振动峰,1033 和945 cm-1是P-O-C的伸缩振动峰,1195、1170和880 cm-1是环三磷腈的P-N伸缩振动峰。对比D2和D1的红外光谱发现,D2中3283 cm-1处出现羟基的特征吸收峰,在2960和2830 cm-1处CH3的伸缩振动峰消失,1033 cm-1处C-O-C的伸缩振动峰消失,表明D1的甲基已完全脱除,形成含羟基的D2的化学结构。对比D3和D2的红外光谱发现,3283 cm-1处羟基的伸缩振动峰已基本消失,新出现了2923、2850和1325 cm-1处CH2的伸缩振动峰、1201和1027 cm-1处 C-O-C的伸缩振动峰、1135 cm-1处C-N-C的伸缩振动峰和970 cm-1处噁嗪环的特征吸收峰,表明D2中的酚羟基已参与成环,形成苯并噁嗪环,证实产物D3确实是磷腈型苯并噁嗪。观察图2 (b) 和(c)可以得出同样的结论,且随着噁嗪环数量的增加,噁嗪环特征吸收峰的强度在加强,在3300-3500 cm-1处可以看到逐渐出现微弱的羟基的吸收峰,可能由于关环不彻底,有微量羟基残留所致。

(a)D1, D2, D3的红外光谱图

(b)T1, T2, T3的红外光谱图

(c)F1, F2, F3的红外光谱图图2 中间体与产物的红外光谱图
产物D3的核磁共振氢谱图如图3(a)所示,δ 4.89(s)对应噁嗪环上Ar-CH2-N的亚甲基质子峰,δ 6.01(s)对应噁嗪环上O-CH2-N的亚甲基质子峰,δ 6.65~6.67(d)和δ 7.01~7.05(m)对应噁嗪中苯环以及与噁嗪环相连苯环上的质子峰,δ 6.53~6.55(d)、δ 7.01~7.04(t)和δ 7.07~7.11(m)分别对应苯氧基上苯环的质子峰,δ 7.26(m)处的峰由所用溶剂CDCl3中微量的CHCl3造成的,δ 7.51(m)处的峰则由苯并噁嗪合成中残余的微量酚羟基造成的。此外根据,δ 4.89~7.11的7个峰面积积分比为2.02:1.91:3.98:1.00:4.99:2.01:6.09,与理论比值4:4:8:2:10:4:12相近,说明氢核的化学位移符合目标分子结构特征。同理图3(b)和3(c)所示的产物T3和F3的1H NMR谱图具有类似化学位移特征,同样可以观察到T3的核磁共振氢谱图中δ 4.56~7.24的5个峰面积积分比为6.08:5.91:17.90:8.99:12.01,与理论比值6:6:18:9:12相近,F3的1H NMR谱图中5个峰面积积分比为8.07:7.99:19.91:10.05:12.01,与理论比值8:8:20:10:12相近,进一步证实产物结构符合预期。

(a)D3的核磁共振氢谱
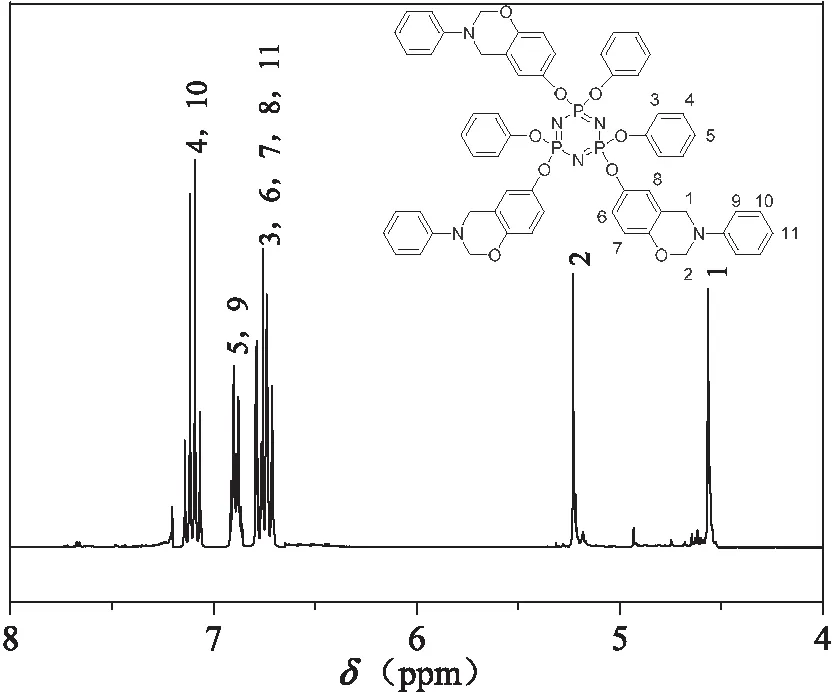
(b)T3的核磁共振氢谱

(c)F3的核磁共振氢谱图3 产物的核磁共振氢谱图
2.2 热性能分析
2.2.1 热固化行为分析
图4(a)为产物D3在不同固化温度下的红外光谱图,在3442 cm-1处酚羟基特征吸收峰的强度随温度升高而加强,反映了苯并噁嗪环在开环聚合过程中酚羟基出现并增多;1325、1201、1027、970 cm-1处的苯并噁嗪环的特征吸收峰的强度随温度升高而逐渐减弱直至消失,说明产物已基本完成开环聚合反应。另外,1033、945、880 cm-1处P-O-C和P-N特征吸收峰随着温度升高未明显变化,说明在苯并噁嗪开环聚合过程中环三磷腈结构未受影响并保持不变。观察图4(b)和(c)可以得出相同的结论。
2.2.2 热稳定性分析
D3、T3和F3的DSC曲线如图5(a)所示,随着噁嗪环数量增加,由D3至F3的开环聚合起始温度明显降低,这可能与分子结构中酚羟基数目有关,多噁嗪环结构的磷腈型苯并噁嗪在合成过程中受空间位阻的影响残留了更多的酚羟基,而酚羟基对开环聚合可起到催化作用[14],因此导致开环聚合起始温度降低。
固化后产物PD3、PT3和PF3的TGA曲线如图5(b)所示,随着噁嗪环数量增加,由D4至F4的固化物起始热分解温度及800℃残炭率明显增高,这可能是由于噁嗪环数量增多,聚合后交联密度增大,提高了聚合物的热稳定性。

(b)在不同固化温度下T3的红外光谱图

(c)在不同固化温度下F3的红外光谱图图4 在不同固化温度下产物的红外光谱图
2.3 阻燃性能分析
表1为固化后产物PD3、PT3、PF3、PBa和PBf的极限氧指数和燃烧测试结果,PD3、PT3和PF3的阻燃等级均可达V-0级,氧指数均高于PBa和PBf,且PD3至PF3,固化物的极限氧指数逐渐增大,这可能由于噁嗪环增加,交联密度增大,磷氮协效增强,提升了固化物阻燃性。

(a)D3~F3的DSC曲线

(b)PD3~PF3的TGA曲线图5 产物的DSC和TGA曲线

表1 固化后产物的LOI和UL-94阻燃等级
3 结论
(1)以六氯环三磷腈为起始原料,通过三步反应设计合成了三种含有不同数量的噁嗪环结构的磷腈型苯并噁嗪,采用红外光谱和核磁氢谱对产物进行表征,证明合成了目标产物。
(2)DSC结果表明,三种产物开环聚合起始温度低于200℃,且随着噁嗪环数量的增加,开环聚合起始温度下降。
(3)TGA结果表明,固化物5%wt的起始热分解温度超过330℃,800℃时残炭率超过55%,随着噁嗪环数量的增加,固化物热分解温度及残炭率增高。
(4)阻燃性结果表明,固化物的极限氧指数超过35%,燃烧等级(UL-94)达V-0级,随着噁嗪环数量的增加,固化物阻燃性升高。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
中国抗生素杂志(2022年7期)2022-08-18
燃料化学学报(2022年5期)2022-05-30
云南化工(2020年11期)2021-01-14
应用化工(2020年9期)2020-09-29
长春师范大学学报(2019年4期)2019-04-29
中国油脂(2019年1期)2019-01-23
合成化学(2015年4期)2016-01-17
海军航空大学学报(2015年1期)2015-11-11
中南民族大学学报(自然科学版)(2014年4期)2014-08-06
