
F掺杂四方钙钛矿结构BiFeO3的铁磁性和电子结构第一性原理研究
2019-05-21 03:52魏丽静郭建新
人工晶体学报 2019年4期
魏丽静,郭建新
(1.华北电力大学科技学院,保定 071051;2.河北大学物理科学与技术学院,保定 071002)
1 引 言
铁酸铋(BiFeO3)是一种多用途的钙钛矿铁电材料[1-3]。作为单相室温多铁材料,它具有高的铁电相变居里温度Tc=1103 K和反铁磁相变奈尔温度TN=643 K,是最具应用前景的磁电材料之一[1]。室温下,BiFeO3体材料呈现畸变钙钛矿的菱形结构,属于R3c空间群,铁电自发极化强度比较微弱,仅有5~10 μC/cm2[2-4]。制备成薄膜后,BiFeO3转变为四方钙钛矿结构,具有巨大的自发极化强度,达到100~150 μC/cm2[5-7]。但其反铁磁性使得BiFeO3的铁磁性很弱,影响了BiFeO3的磁电耦合效果,极大的限制了BiFeO3的实际应用。因此,提高BiFeO3的铁磁性对其应用具有十分重要的意义。通常,替位掺杂是一种有效的实验手段。科研人员使用Dy替代Bi,掺杂后结构的扭曲导致部分自旋结构被抑制,打破了Fe3+的反铁磁平衡,进而获得磁矩的增加[8]。也有研究人员使用非磁性金属原子Mg替换磁性原子Fe[9],BiFeO3中原来的磁矩平衡被打破,使体系的磁性明显增加。除Bi位和Fe位的替换掺杂外,采用非金属原子替换BiFeO3的O位也是一种可选择的途径[10]。F作为第七主族元素,具有较高的电负性,化合价态为-1价,用来替代O原子,可以使Fe的价态发生改变,来调节BiFeO3的磁距。实验上,Hu等使用溶胶-凝胶法在BiFeO3中掺入不同比例的F形成BiFeO3-xFx[10]。结果发现,在x=0.25时,其铁磁性比未掺杂时提高了两个量级,F的比例对BiFeO3-xFx的影响至关重要。然而,该工作基于R3c空间群的菱方畸变钙钛矿的BiFeO3,此时的BiFeO3的极化强度比较小。四方结构的BiFeO3(T-BiFeO3)具有较大的极化强度,本文基于这种结构,采用第一性原理方法深入分析F掺杂后的BiFeO3的结构变化和磁矩大小,从微观上理解F掺杂提高的铁磁性的T-BiFeO3机制,为实验上制备出具有较好磁电耦合性能的多铁材料打下基础。

图1 F的掺杂位置以及T-BiFeO3铁磁序示意图(向上和向下箭头分别表示上自旋和下自旋)(a)Sa site;(b)Sb site;(c)A-type;(d)C-type;(e)G-type;(f)F-typeFig.1 Sketch map of F doped sites and different magnetic ordering(the up and down arrows represent up and down spins, respectively)
2 计算方法和理论模型
本文所有计算采用基于密度泛函理论的第一性原理计算软件VASP程序包[11]。电子相互作用势采用投影缀加波(PAW)方法来描述[12],交换关联泛函用广义梯度近似(GGA)的Perdew-Burke-Ernzerhof (PBE)来处理[13]。所有计算模型采用平面波基矢来展开波函数,且平面波的截断能设置为500 eV,所有原子和晶胞弛豫的收敛精度为10-5eV,原子受力小于0.2 eV/nm。第一布里渊区的k点网格采用Monkhorst-Pack方案[14]自动生成。由于Fe为过渡金属磁性原子,一般的GGA方法不能准确处理其氧化物强关联磁性体系,因此,本文的计算全部采用GGA+U方法[15],且U值采用其有效值Ueff(Ueff=U-J)为 4 eV[5]。模型采用BiFeO3四方钙钛矿结构,考虑BiFeO3的反铁磁性,晶胞采用2×2×2的超胞结构,且使用3×3×3大小的Monkhorst-Pack方案自动生成的k点网格。如图1,计算模型考虑到两种F掺杂位置(Sa和Sb)和T-BiFeO3的三种反铁磁序(A-type,C-type和G-type)和一种铁磁序(F-type)。图2(a)是F的两种掺杂位置分别在T-BiFeO3的四种磁序时的总能。可以看出,在同种磁序结构情况下, F替代O的Sb位都是T-BiFeO3最稳定的结构,能量与Sa位相比要低0.34 eV。因此可以推断,F掺杂BiFeO3时优先依次替代Sb位O的位置(图1(b))。由图2(a)还可以看出,F掺杂后C-type为体系的基态结构,比G-type的BiFeO3的能量要低0.05 eV/formula,而A-type和F-type的能量相对要高,因此接下来的计算都基于C-type型磁结构T-BiFeO3。
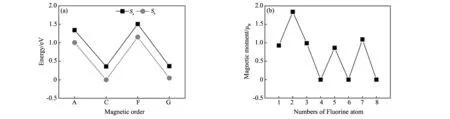
图2 (a)F的两种掺杂位置分别在BiFeO3的四种磁序时的超胞总能(Sb位置掺杂的C-type的总能作为能量参考零点);(b)F的不同掺杂比例下的总磁矩Fig.2 (a)Total energies of BiFeO3supercell with different magnetic ordering for two F doped sites(energies areplotted relative to the C-type BiFeO3with Sb doping);(b)total magnetic moment of different F atoms
3 结果与讨论
3.1 掺杂体系的磁性
为了判断F掺杂的难易程度,通过下面的公式计算了F掺杂T-BiFeO3的形成能:Ef=Edoped-Epristine-μ0-μF,其中Edoped和Epristine分别为掺杂和未掺杂的T-BiFeO3总能,μ0和μF则为O和F的化学势,它们都来自单个原子的总能。形成能越小则表示越容易进行掺杂,掺杂后的结构则越稳定。如果形成能为负值,则表示掺杂可以自发完成。通过上述公式得到的F替代O的形成能为1.55 eV,表示F的替代掺杂需要外界提供一定的能量才可以完成。但众所周知,BiFeO3薄膜制备过程中,在缺氧条件下很容易形成氧空位,如果此时通入适量的F,F可以直接填充到O空位上,从而实现F掺杂到BiFeO3基体中。另外,也可以通过制备出含F的陶瓷靶材实现F的掺杂[10]。
图2(b)中给出了在一个T-BiFeO3超胞中,当增加F占比时,根据总能最小原理计算得到的O被替代顺序。在一个超胞中替代F的数目不同,体系的总磁矩也不同,当F的个数为2的时候,体系的总磁矩为最大,达到2 μB。F的增加并不一定增加体系的总磁矩,相反,有时还会恢复到原来体系的反铁磁性,这跟F的替代位置密切相关。
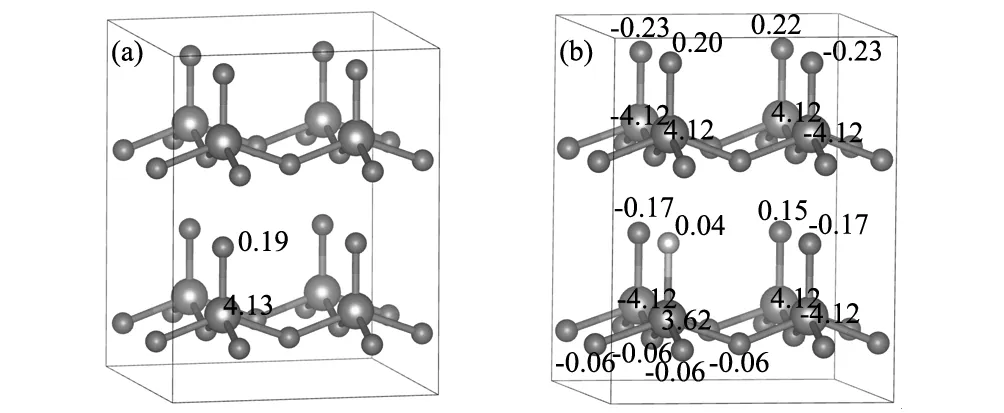
图3 (a)掺杂F前各个原子的磁矩;(b)掺杂F后各个原子的磁矩Fig.3 (a)Magnetic moment of every atoms without F doped;(b)magnetic moment of every atoms with F doped
为了分析简单,以F替代一个O的位置为例来分析F掺杂T-BiFeO3的铁磁性来源。图3给出了一个F掺杂前后,T-BiFeO3超胞中各个原子的磁矩。可以看出,未掺杂的T-BiFeO3的除了Fe原子具有一定的磁矩外,Sb位的O原子也具有一定的磁矩。当F替代O后,其它Sb位的O原子也发生了相应的变化,与F近邻的Sb位O原子变化更加明显,但由于磁距方向相反,Sb位的O总体上对系统总磁矩贡献不大。F最近邻的Fe原子(Fe')的磁矩变化最明显,由未掺杂的4.128 μB 变为掺杂后的3.62 μB。F的磁矩为0.04 μB,与原来O的磁矩0.19 μB相比,相差0.15 μB。另外,与Fe'相邻的氧原子也由原来无磁矩变为有磁矩,对系统磁矩产生贡献。综上所述,系统磁矩的增加主要来源于F,Fe'及其Fe'最近邻的Sa位O。这主要是因为F和O相差一个电子,与Fe的成键方式发生了变化,改变了原来的自旋占据态,影响了Fe及其周围O原子的原子磁矩,体系呈现出一定的净磁矩。Bader电荷分析[16]表明,与掺杂之前相比,F最近邻Fe的外层电子多了0.235个电子,Fe的化合价态降低,体系呈现出净磁矩,这与实验上的F掺杂BiFeO3出现宏观的整体磁矩是一致的[10]。F掺杂后,所产生磁矩的方向与原来Fe'和Sb位O的磁矩的方向密切相关,所产生的磁矩方向与之相反。
F的掺杂数目为2时,T-BiFeO3表现为最大的整体净磁矩。虽然存在结构上的差异(实验上为菱方结构),但实验上同样发现,在x=0.25时,BiFeO3-xFx展现最大的宏观磁矩。这是因为第二个F替代O的位置不仅在能量上使得体系具有最小的总能,达到最稳定的结构,而且F所占据的位置与第一个F占据的位置一样,以同样原理对体系贡献了磁矩。但随着F的增加,各个F所占据的位置对体系磁矩的贡献相互抵消,致使最终体系的总磁矩不会更大。通过对掺杂前后Bi的磁矩进行计算可以发现,Bi对体系磁矩没有贡献。
3.2 掺杂后体系的电子特性
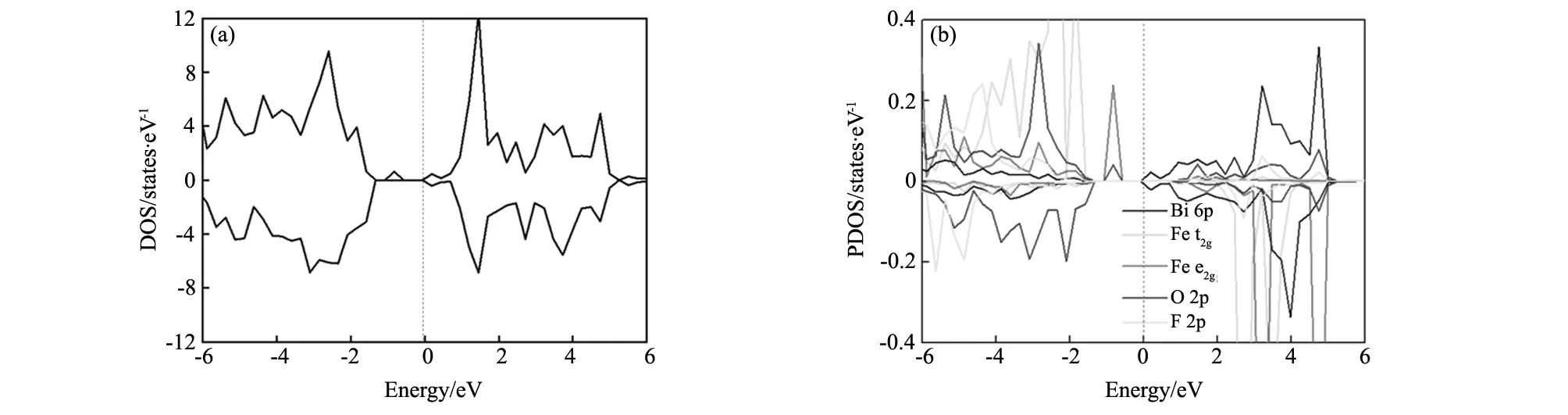
图4 掺杂后F所在单胞的态密度图 (a)总态密度图;(b)各个原子的投影态密度图Fig.4 Density of states of the unit cell T-BiFeO3with F doped(a)total density of states;(b)projected total density of states for every atoms
为了进一步研究F掺杂对T-BiFeO3的电子特性的影响,仍然以掺杂1个F的C-type型为例,给出F掺杂后的总态密度图和F所在的铁氧八面体的Bi、Fe、O的相关轨道的分波态密度图,如图4所示。从图4(a)总态密度图可以看出,掺F后,T-BiFeO3仍然保持半导体性质,但上自旋和下自旋的半导体带隙并不相同,这主要因为F掺杂后改变了体系的磁性,上自旋和下自旋不平衡造成的。从图4(b)中可以看出,上自旋的价带顶主要来自于Fe的e2g轨道带顶主要来自于Fe的e2g轨道和O的2p轨道,而F则没有贡献。图5(a,b)给出了组成Fe e2g的两个轨道3dz2和3dx2-y2的态密度图和O 2px、2py和2pz的态密度图。可以看出,3dx2-y2轨道对于上自旋价带顶的贡献最大,这主要是因为F比O少提供了一个电子与Fe成键,Fe多余的电子与其它周围的O原子(Sa位置的O)产生相互作用,使得体系的总体能量升高。为了能够更形象的表征Fe与F和O的成键情况,使用第一性原理做了包含四个Fe原子的BiFeO3(100)面电子局域函数(ELF),如图6所示。ELF反映了电子在晶体结构中的局域化程度和相对化学键强度,可以用下面数据公式进行描述[17-18]:
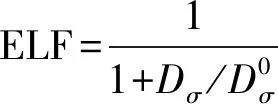
(1)
磁电耦合除要求具有较大的磁化强度外,还要有较大的极化强度,对菱方BiFeO3,F的掺杂使原来非常小的极化强度(0.12 μC/cm2)增加到5.1 μC/cm2[10]。而对于本文研究的具有最大磁矩的T-BiFeO2.75F0.25,未掺杂F的时候具有非常大的极化强度(~150 μC/cm2)[19]。通过berry相方法[20],计算得到的极化强度为85.7 μC/cm2。与未掺杂相比,虽然下降了很多,但其极化强度仍然很大,这对磁电耦合效应非常有利。
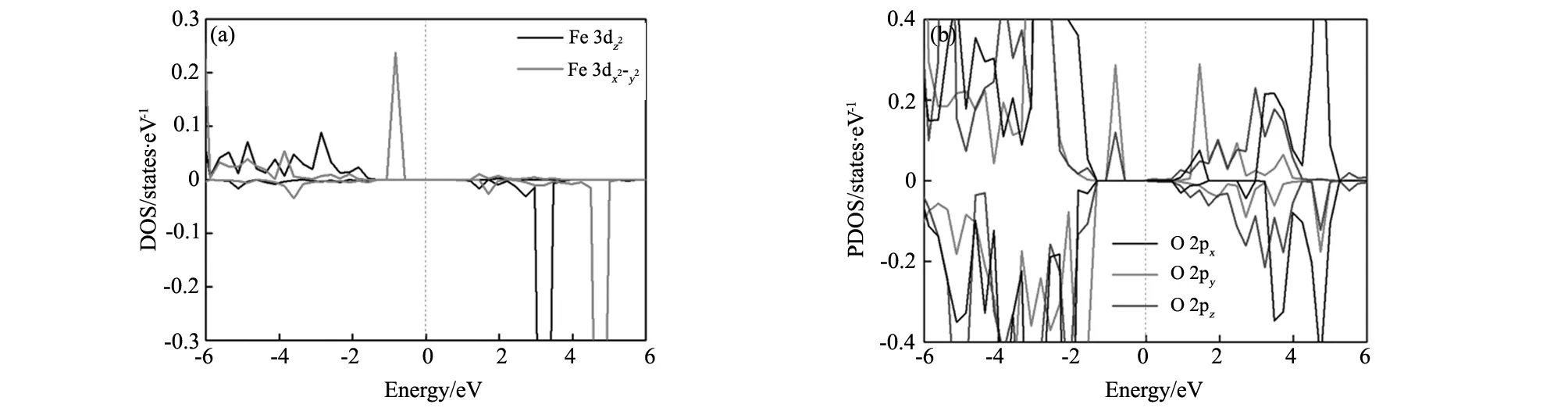
图5 掺杂后F所在单胞的T-BiFeO3投影态密度图 (a)F;(b)OFig.5 Projected density of states of of T-BiFeO3with F doped Fe(a) and O(b) for the unit cell

图6 包括四个Fe的T-BiFeO3(100)面ELF图Fig.6 ELF map of T-BiFeO3(100) containing four Fe atoms
4 结 论
本文计算了F替代O的不同掺杂位置和T-BiFeO3的不同磁序(三种反铁磁序和一种铁磁序)的总能。结果发现,F占据Sb位置的C-type反铁磁序T-BiFeO3具有最小的能量,为最稳定结构,是掺F的T-BiFeO3的基态。F掺杂T-BiFeO3总磁矩改变的大小与F的掺杂浓度相关。当掺杂浓度x=0.25时,T- BiFeO2.75F0.25的总磁矩最大,这是因为F掺杂的浓度不同,F所替代的O的位置也不同,而F的占位直接影响了总磁矩的大小。进一步的态密度分析表明,F的掺入造成了T-BiFeO3的整体自旋态的不平衡,特别是F最近邻的Fe与周围的Sa位O杂化在费米能级附近形成了杂质态。电子局域函数的分析表明,不同于Fe与O所形成的金属键和离子键的混合键,Fe与F的成键更接近离子键。通过berry相方法计算得到的掺F后T-BiFeO2.75O0.25仍具有很大的极化强度,达到85.7 μC/cm2。说明合适浓度F的掺杂对T-BiFeO3的磁电耦合具有积极影响。
猜你喜欢
现代财经-天津财经大学学报(2022年5期)2022-06-01
航天电子对抗(2022年2期)2022-05-24
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
北京航空航天大学学报(2021年9期)2021-11-02
军民两用技术与产品(2020年2期)2020-03-14
航天电子对抗(2019年4期)2019-06-02
学生天地·小学低年级版(2016年9期)2016-05-14
