多黏菌素抗菌机理及耐药机制研究进展
2019-07-25 00:38乔涵游雪甫李聪然
中国抗生素杂志 2019年7期
乔涵 游雪甫 李聪然
(中国医学科学院北京协和医学院医药生物技术研究所,抗感染药物研究北京市重点实验室,北京 100050)
临床中抗生素的过度使用对细菌施加了强烈的选择压力,加速细菌产生抗生素耐药(antimicrobial resistance,AMR)。近年来,细菌耐药问题,特别是多黏菌素耐药革兰阴性菌引起的感染,越来越受到人们的广泛关注。世界卫生组织的有关资料显示,全球每年约有70万人死于抗生素耐药菌引发的感染[1]。多黏菌素类抗生素是治疗多重耐药革兰阴性菌感染的最后一道防线,若治疗细菌感染的最后一道防线也被细菌打破,人类将有可能进入前抗生素时代。
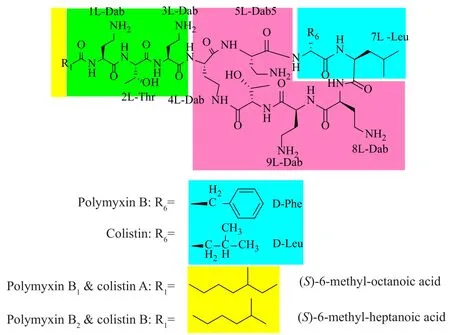
图1 多黏菌素化学结构[5]Fig.1 Chemical structure of polymyxins[5]
多黏菌素于1947年被发现,是一种古老的药物,价格低廉,它是由多黏芽孢杆菌产生的一组有A~E等组分的环肽类抗生素[2]。目前用于临床的产品主要是多黏菌素B和多黏菌素E的硫酸盐和甲磺酸盐。多黏菌素由线性三肽部分连接N-脂肪酰基链和环状七肽构成,4-位L-Dab(α,γ-二氨基丁酸)与10-位L-Thr缩合形成七肽环。多黏菌素B和多黏菌素E的结构十分相似,仅在6-位氨基酸上存在差别。多黏菌素B的6-位是D-Phe,多黏菌素E的6-位是D-Leu(图1)[3]。多黏菌素对大多数肠杆菌科细菌有活性,包括肠杆菌属,克雷伯菌属,柠檬酸杆菌属,沙门菌属和志贺菌属等,对常见非发酵性革兰阴性菌也有显著活性,包括鲍曼不动杆菌,铜绿假单胞菌和嗜麦芽寡养单胞菌[4]。但一些菌种对多黏菌素天然具有抗性,包括变形菌属、摩氏摩根菌、普罗威登斯菌属、黏质沙雷菌、马来假单胞菌、洋葱伯克霍尔德菌、色杆菌属、爱德华菌属、布鲁菌、军团菌、弯曲菌和霍乱弧菌。多黏菌素对革兰阴性球菌(奈瑟球菌属),革兰阳性菌和厌氧菌没有活性[4]。因为多黏菌素抗菌谱窄,毒性大,所以最初应用于兽药和饲料添加剂。后来随着碳青霉烯类抗生素耐药菌的产生,多黏菌素也开始渐渐应用于临床治疗,并被誉为临床治疗多重耐药革兰阴性菌感染的最后一道防线。随着多黏菌素应用的增多,有些细菌对其产生了耐药性,而多黏菌素耐药机制也成为了研究热点。多黏菌素耐药的完整机制尚未完全阐明,但多黏菌素耐药现象的出现很可能与用药时间的延长及用药量的加大有关。本文将从多黏菌素的抗菌机理、副作用产生机制、耐药机制等方面进行综述和讨论。
1 多黏菌素抗菌作用机制
革兰阴性细菌外膜(outer membrane,OM)的关键作用是作为渗透屏障[6]。多黏菌素的原始靶标是外膜上的脂多糖(lipopoly saccharide,LPS)组分。LPS由3个结构域组成,即脂质A、核心多糖和O-抗原。脂质A充当疏水端,脂肪酰基链的紧密堆积有助于稳定整个外膜的结构[7]。多黏菌素通过与LPS的脂质A组分相互作用使细菌外膜通透性增加(图2)来发挥它们的抗菌作用。多黏菌素的抗菌谱之所以窄,也是因为其只能与有限几种LPS结合导致[6,8-9]。虽然LPS是最初的靶标,但多黏菌素的确切作用方式仍然不清楚,目前已知的机制有以下几种。
1.1 “自促摄取”机制
该机制是一个被广为认可的模型,该模型认为多黏菌素两性性质对多黏菌素分子通过外膜屏障至关重要。在这个模型中,多黏菌素Dab残基上游离的氨基在生理条件下发生质子化作用,并与类质A磷酸基阴离子(图3)产生静电吸引作用。质子化的多黏菌素取代二价阳离子(Mg2+和Ca2+)来稳定脂多糖层,多黏菌素分子的N-脂肪酰基链和6、7位疏水部分的插入,弱化了相邻脂质A脂肪酰基链的堆积,导致外膜单层的扩展,最终使外膜膨胀[10-11]。随后,多黏菌素介导的外膜和细胞膜融合通过诱导磷脂交换实现,最终导致渗透失衡和细胞死亡[5]。
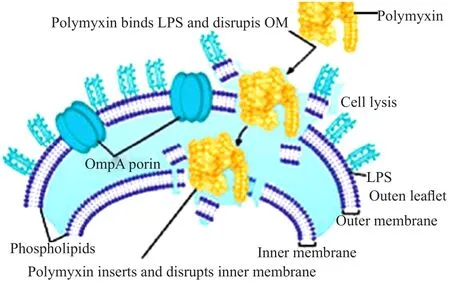
图2 多黏菌素作用于LPS,增加细菌外膜的通透性[5]Fig.2 Polymyxin interacts with LPS and permeabilizes the outer membrane of bacteria[5]

图3 多黏菌素B与细菌外膜相互作用示意图[11]Fig.3 Interaction of polymyxin B with bacterial outer membrane[11]
1.2 多黏菌素介导的内膜和外膜的囊泡之间的接触
这一假设是基于多黏菌素B结合于阴离子磷脂囊泡,能够形成囊泡-囊泡接触的实验结果[11-12]。这些接触能够促进囊泡之间的磷脂交换。在革兰阴性菌中,多黏菌素B在周质空间中可以形成内膜和外膜之间的接触和脂质交换,而内外膜都富含磷脂酰甘油和心磷脂,由此产生的磷脂交换会降低其成分的差异,从而导致细胞渗透不平衡和细胞活力的丧失[12]。
1.3 抑制细菌内膜中重要的呼吸酶
一般来说细菌呼吸链由3个复合体组成,醌和还原型辅酶(NADH)作为在大蛋白质复合物之间穿梭电子和质子的载体来发挥作用[13]。在复合物1中,已鉴定出NADH氧化酶家族的3种内膜呼吸酶:NDH-1、NDH-2和钠转运NADH-Q氧化还原酶[13-14]。多黏菌素通过羟基自由基的释放浓度依赖性地抑制革兰阴性菌内膜中NDH-2酶活性,进而诱导鲍曼不动杆菌和其他革兰阴性菌的快速杀灭[15]。
1.4 抗内毒素作用
除直接抗菌活性外,多黏菌素还具有有效的抗内毒素活性。革兰阴性细菌的内毒素是LPS分子的脂质A。多黏菌素B结合并中和LPS的脂质A,阻断了LPS和完整细胞在孵育后诱导内毒素休克介质肿瘤坏死因子(TNF)的能力,从而有效保护了被革兰阴性菌所感染的宿主[16-17]。
2 多黏菌素的副作用机制
虽然多黏菌素对某些耐药的革兰阴性菌感染有着突出且不可替代的作用,但因为多黏菌素抗菌谱窄,毒性大等问题还是不能成为一个很理想的药物。随着碳青霉烯类抗生素耐药菌的产生,多黏菌素不得不成为对抗多重耐药的革兰阴性菌的最后一道防线,但其显著的副作用包括神经毒性和肾毒性也是不可忽视的问题。因此,多黏菌素临床使用时产生副作用的相关机制很有必要进行了解和探讨。
2.1 神经毒性
神经毒性表现为头晕、嗜睡、面部和周围组织感觉异常、眩晕、视力下降、共济失调和神经肌肉接头阻滞,可能导致呼吸衰竭或呼吸暂停,神经毒性是剂量依赖性和可逆性的。分子伴侣HSP90是活细胞中必不可少的蛋白质之一[18-19]。黏菌素对HSP90的ATP酶活性没有影响,且低浓度不影响HSP90的分子伴侣活性[20],但黏菌素可特异性结合HSP90,通过N-端结构域诱导HSP90聚集,故学者们推测神经毒性是由于大脑中高浓度的黏菌素与HSP90的聚集,导致HSP90的失活,而HSP90的失活可能导致细胞功能的崩溃,并且可以间接诱导细胞凋亡。
2.2 肾毒性
肾毒性是最常见的不良反应,特别是对于新推荐的高剂量治疗方案。肾毒性主要包括急性肾小管坏死,表现为肌酐清除率降低,血清尿素和肌酐水平升高。与神经毒性类似,肾毒性也是剂量依赖型的,且在早期停药症状可逆。黏菌素的肾毒性主要与其D-氨基丁酸和脂肪酸成分有关,肾毒性机制类似于它的抗菌作用:(1)细胞膜毒性:黏菌素增加了肾小管上皮细胞膜的通透性,导致阳离子、阴离子和水分流入,引起细胞肿胀和细胞溶解[21]。(2)氧化应激损伤:指机体在需要清除体内老化的细胞,或在遭受各种有害刺激时,体内高活性分子,如活性氧自由基(ROS)和活性氮(RNS)自由基产生过多,氧化程度超出氧化物的清除,氧化系统和抗氧化系统失衡,从而导致组织损伤[22]。维生素C、褪黑素及N-乙酰半胱氨酸等抗氧化剂可保护黏菌素诱发的肾细胞脂质过氧化及凋亡等副作用发生[23]。(3)细胞凋亡途径(即线粒体,死亡受体和内质网途径)和自噬途径:动物及体外细胞模型研究证实,线粒体凋亡通路、内质网凋亡通路及死亡受体凋亡通路参与黏菌素诱导的细胞凋亡,且线粒体及死亡受体通路为黏菌素诱导细胞凋亡的主要通路[24-25]。(4)另外,黏菌素还可能通过抑制宿主核质和核糖体的功能糖体功能产生肾毒性。
2.3 其他毒性
黏菌素的其他不良反应包括过敏反应、皮疹、荨麻疹、全身性瘙痒、发烧和轻度胃肠道疾病。据报道,黏菌素引起的过敏反应发生率约为2%[26]。此外,伪膜性结肠炎代表了黏菌素的另一副作用,尽管罕见,但也是黏菌素治疗的潜在副作用。使用雾化黏菌素治疗可能进一步加重支气管收缩和胸闷。然而,在开始用雾化黏菌素治疗之前吸入β2激动剂治疗可以预防支气管收缩的发展。脑室内给予黏菌素,特别是高剂量,可能导致惊厥。但这些不良反应大多比较少见,故其发生机制还有待研究。
3 多黏菌素耐药机制
随着多黏菌素耐药现象的出现和增加,耐药机制的研究已成为目前的一项热点问题。多黏菌素耐药的完整机制尚未完全阐明,目前已明确的主要有以下4种[27]:对外膜脂多糖(LPS)结构进行修饰改造;屏障系统或广谱外排泵系统的活化;存在药物的降解蛋白;细菌的异质性耐药等[11,28-29]。
3.1 对外膜脂多糖(LPS)结构进行修饰改造
多黏菌素对革兰阴性菌作用的关键第一步是多黏菌素带正电的α,γ-二氨基丁酸(Dab)残基和脂质A上的带负电荷的磷酸基团之间的静电相互作用[3,5]。因此,许多细菌对多黏菌素耐药的机制是基于降低这种初始静电相互作用的脂质A头部基团的修饰。在大肠埃希菌、沙门菌血清型鼠伤寒沙门菌、肺炎克雷伯菌和铜绿假单胞菌中,用带正电荷的基团,例如4-氨基-4-脱氧-L-阿拉伯糖和/或磷酸乙醇胺修饰脂质A的磷酸盐,可减少净负电荷量而介导菌株的多黏菌素耐药性(图4)。这种修饰脂质A的机制又分为质粒介导和染色体介导两种。
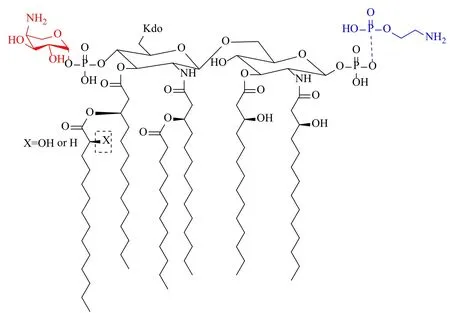
图4 多黏菌素Dab残基与脂质A产生静电吸引作用[3]Fig.4 Polymyxin Dab residue and lipid A produce electrostatic attraction[3]
3.1.1 质粒介导的多黏菌素耐药机制
过去认为,黏菌素的耐药机制是由染色体突变介导,并不能通过基因的水平转移引起耐药,但2015年首次在动物源大肠埃希菌(Escherichia coli)中发现了质粒介导的黏菌素耐药基因mcr-1(mobile colistin resistance)[30],并通过体外接合试验证明了该基因可通过可接合性质粒在不同菌种间传播,介导低水平的黏菌素耐药。
mcr-1基因全长1626bp,GC含量49%,位于IncI2型质粒上。产物MCR-1蛋白长度为541个氨基酸。氨基酸序列分析结果显示MCR-1与之前报道的磷酸乙醇胺转移酶家族有60%左右的相似性,与类芽孢杆菌编码的磷酸乙醇胺转移酶的同源性达63%[30]。
磷酸乙醇胺转移酶可将磷酸乙醇胺基团(PEtN)添加到脂多糖表面的类脂A上,从而降低黏菌素与脂多糖的静电作用,导致细菌对黏菌素耐药[31-32]。通过ESI-MS分析,证实MCR-1具有磷酸乙醇胺转移酶活性,即可催化磷酸乙醇胺与脂多糖表面类脂A 结合,介导黏菌素耐药[30]。通过对MCR-1蛋白模拟结构分析,MCR-1蛋白是一个膜结合蛋白,N端为膜结合域,含有5个疏水的跨膜α-螺旋结构,C端为亲水结构域。跨膜结构域能固定MCR-1于细胞内膜的周质面,在细胞周质中完成对类脂A的共价修饰,同时指出MCR-1中的5个氨基酸残基(E246、T285、H395、D465和H466)可能与其底物结合活性相关[33]。MCR-1与两个已知的可溶性磷酸乙醇转移酶结构(图5)相似,分别为脑膜炎奈瑟菌的LptA和空肠弯曲杆菌的EptC[30]。

图5 MCR-1蛋白结构模型[33]Fig.5 MCR-1 protein structure model[33]
除mcr-1外,2016年6月在比利时大肠埃希菌中鉴定出一种新型质粒介导的黏菌素抗性基因mcr-2[34]。mcr-2基因长1617bp,比mcr-1短9个碱基,与mcr-1具有76.75%的同源性。MCR-1和MCR-2蛋白显示80.65%的同源性。2017年4月,第三个移动黏菌素抗性基因mcr-3被发现[35]。mcr-3基因全长1626bp,分别与mcr-1和mcr-2显示45.0%和47.0%的核苷酸序列同源性。MCR-3的氨基酸序列分别与MCR-1和MCR-2具有32.5%和31.7%的氨基酸同源性。2017年在mcr-3发现不久,mcr-4也被发现,其基因全长1820bp[36]。2017年12月,有被称为mcr-5(1644bp)的基因被发现[37],它是Tn3家族的7337bp转座子的一部分,并且通常位于相关的多拷贝ColE型质粒上。mcr-5与mcr-1和mcr-2有81.23%的氨基酸序列同源性并且被认为都起源于莫拉菌属。而MCR-5与MCR-1、MCR-2、MCR-3和MCR-4的蛋白质序列同源性分别为36.11%、35.29%、34.72%和33.71%[37]。
3.1.2 染色体介导的多黏菌素耐药机制
对于奇异变形菌和黏质沙雷菌,对多黏菌素的天然耐药与磷酸乙醇胺(pEtN)或4-氨基阿拉伯糖(L-Ara4N)阳离子基团修饰LPS的脂质A有关。这种修饰增加了多黏菌素初始靶标LPS的电荷,因此降低了多黏菌素的结合,导致这些菌株的固有耐药[38-40]。
对于肠杆菌科细菌,也是向LPS添加阳离子基团(如L-Ara4N和pEtN),与其有关的基因包括编码直接参与LPS修饰的酶基因和操纵子(负责合成阳离子基团和/或将其添加到LPS)。如(1)pmrC基因:pmrCAB操纵子编码3种蛋白质,即磷酸乙醇胺转移酶PmrC,反应调节剂PmrA(也称为BasR)和传感器激酶蛋白PmrB(也称为BasS)[41]。(2)pmrE基因和pmrHFIJKLM操纵子(也称arnBCADTEF或pbgPE操纵子):负责L-氨基阿拉伯糖基团(L-Ara4N)的合成及其对脂质A的固定[42]。(3)pmrA和pmrB基因:PmrB是具有组氨酸激酶活性的蛋白质,其通过磷酸化来激活PmrA。PmrA依次激活参与LPS修饰的pmrCAB操纵子,pmrHFIJKLM操纵子和pmrE基因的转录[41]。(4)phoP和phoQ基因:PhoQ是一种具有组氨酸激酶活性的蛋白质,通过磷酸化活化PhoP。PhoP反过来激活pmrHFIJKLM操纵子的转录,参与将L-Ara4N添加到LPS的脂质A上[43-44]。PhoP也可以通过PmrD连接蛋白直接或间接激活PmrA蛋白,导致pEtN添加到LPS中。(5)mgrB基因:MgrB(也称为YobG)是47个氨基酸的小跨膜蛋白。PhoP激活后,mgrB基因上调。MgrB蛋白抑制PhoQ编码基因的表达,因此MgrB蛋白对phoPQ操纵子有负反馈作用。而mgrB基因的失活导致phoPQ操纵子的过表达,进而引起pmrHFIJKLM操纵子活化,导致L-Ara4N修饰LPS作用而产生耐药[45-46]。(6)crrAB操纵子:crrAB操纵子编码两种蛋白质,调节蛋白CrrA和传感器蛋白激酶CrrB。crrB基因的失活导致pmrAB操纵子的过表达,从而导致pmrHFIJKLM操纵子及pmrC和pmrE基因的激活,最终导致细菌获得黏菌素耐药性。CrrB失活也可能通过激活糖基转移酶样蛋白来修饰脂质A[47-48]。(7)ParRS、ColRS和CprRS 3种双组分体系:ParRS(多黏菌素适应性耐药)双组分体系与多黏菌素的适应性耐药有关[49]。该系统突变导致pmrHFIJKLM操纵子的组成型表达,并因此导致L-Ara4N添加至LPS导致黏菌素耐药。ColRS和CprRS系统的作用可能通过激活phoQ基因或通过其他基因导致黏菌素耐药[50]。
3.2 屏障系统改变或广谱外排泵系统的活化
除对LPS的修饰作用外,还有一些其他机制,如(1)荚膜多糖(CPS)产生增加:一项研究表明,CPS可作为肺炎克雷伯菌抗多黏菌素的保护屏障[51],CPS在细菌表面释放阴离子,这些阴离子与多黏菌素结合,从而减少多黏菌素与LPS的结合,使细菌菌体受到保护[52],产生多黏菌素耐药。(2)内在调节器RamA:它调节与渗透性障碍相关的基因,因此可能参与降低对抗生素的易感性。这种调节因子水平的升高引起LPS的改变,从而降低了对多黏菌素的易感性[53]。(3)膜孔蛋白的作用:ydeI基因编码14kDa的寡糖/寡核苷酸结合折叠蛋白(OB-折叠蛋白),这种OB-折叠蛋白可与膜孔蛋白(OmpD/NmpC:三聚体β-折叠外膜孔蛋白家族成员)相互作用从而增加细菌对多黏菌素的抗性,如肠道沙门菌对多黏菌素的抗性[54]。(4)外排泵的作用:外排泵的编码组分kpnEF和acrAB的突变可导致黏菌素MIC显著降低[55-56]。在低剂量外排泵抑制剂羰基氰化间氯苯腙(CCCP)中培养耐药菌,可降低耐药菌株的MIC(减少128~512倍),并部分或完全抑制抗性亚群的再生[57]。(5)脂质A生物合成基因(lpxA、lpxC和lpxD基因)改变:这些基因可能被插入序列如ISAba11所替换、截断、移码或插入失活,引起的LPS完全丧失,最终导致多黏菌素耐药[58-59]。
3.3 存在药物的降解蛋白
除以上机制外,还有一种直接的耐药机制,就是细菌能够产生降解多黏菌素的酶,从而降解多黏菌素,使其不能与细菌的脂质A相互作用。由于多黏菌素本身属于一种多肽类抗生素,因此能够将其降解的也应是蛋白酶。2018年学者们从若干蛋白酶产生菌中筛选得到高效降解黏菌素的地衣芽孢杆菌DC-01[60]。该菌株可在23~44℃之间产生黏菌素降解酶,但由于未做进一步确证,还不知编码该蛋白酶的基因位于质粒还是染色体中,但仍应该提高警惕以防一些革兰阴性菌获得生产该酶的能力从而引起黏菌素耐药。
3.4 细菌的异质性耐药
异质性耐药是细菌耐药的一种特殊类型,说的是一种表面上同源的细菌,表现出对特定抗生素部分敏感的现象[61],即在体外的常规药敏试验中,菌株表现为敏感,如若改用特殊方法检测,则可以发现细胞的大部分亚群属于敏感,但有一小部分亚群属于耐药,极少数亚群甚至表现为高水平耐药,这部分耐药亚群可以导致临床抗生素治疗失败[62]。(1)鲍曼不动杆菌对黏菌素异质性耐药的机制可能与LPS或/和PmrAB双组分系统的丧失有关[63]。(2)洋葱伯克霍尔德菌对多黏菌素B的异质性耐药取决于盐酸二丁胺分泌水平和YceI的不同。由于盐酸二丁胺具有挥发性,因此也可以以挥发性介导的方式将抗性传递给物理分离的细菌[64]。(3)铜绿假单胞菌对多黏菌素的异质性耐药也与多胺有关[65]。
4 总结与展望
多黏菌素到目前为止还是临床治疗多重耐药革兰阴性菌感染的最后一道防线。因此探索多黏菌素的作用机制和副作用机制,以开发更加有效且副作用更小的多黏菌素类似物;探索多黏菌素的耐药机制,以发现更有效的对抗耐药的靶点都是极为必要的。本文中阐述的机制希望可以对临床治疗和基础探究以及对抗多黏菌素耐药起到部分指导作用。
猜你喜欢
中国合理用药探索(2022年8期)2022-11-26
中国免疫学杂志(2022年10期)2022-07-21
系统医学(2022年5期)2022-06-14
农业与技术(2021年21期)2021-11-17
临床肾脏病杂志(2020年2期)2020-12-12
临床骨科杂志(2020年4期)2020-09-07
中国感染与化疗杂志(2019年4期)2019-01-06
环球时报(2018-02-02)2018-02-02
科学种养(2017年9期)2017-09-12
爱你(2017年17期)2017-06-10