
哌啶在MoP表面C—N键断裂反应实验设计
2019-10-15 06:09朱后禹匙玉华赵联明郭文跃
实验室研究与探索 2019年9期
朱后禹, 匙玉华, 赵联明, 徐 静, 郭文跃
(中国石油大学(华东) 材料科学与工程学院,山东 青岛 266580)
0 引 言
近年来,计算机模拟技术得到了飞速的发展,许多材料的性质可以在其制备和表征前通过计算模拟的方法预测出来。因此,材料研究已经从传统的试错法逐步走向理性的筛选与设计。考虑到材料的应用环境日益复杂,其性能的实验室研究也会变得越发困难,仅依靠传统的实验本身已难以满足新型材料的研发要求。通过计算机模拟可以初步了解超高温、超高压等极端条件下材料的使用性能,基于所计算的材料性能演变规律和机理,可以为进一步改善材料性能提供理论指导。在现代材料学的研究中,计算机“实验”已成为与传统实验具有同等地位的技术手段[1]。对于原子电子层面的问题,一般基于密度泛函理论(Density functional theory,DFT)方法。石油炼制过程中,加氢脱氮(HDN)是除去原油馏分中含氮化合物的重要步骤。含氮化合物会严重抑制HDS反应的进行,因而HDN技术及其催化剂材料是获得超低含硫油品的关键[2-5]。本文尝试对HDN反应过程进行计算实验设计,借助Materials Studio软件程序中的DMol3模块展开计算。实验采用DFT计算方法,依托超算中心的硬件资源,通过模拟确认了哌啶在MoP表面上的吸附构型、能量及C—N键断裂的反应机理。
1 实验设计
加氢脱氮过程一般包括不饱和含氮化合物的加氢、杂环C—N键的断裂以及氨基的脱除,其中,C—N键断裂是整个反应的关键。目前,针对C—N键的断裂机理,人们进行了大量的研究并提出了多种机理,但比较认同的主要是霍夫曼β-H消去机理(E1和E2)和亲核取代机理(SN1和SN2)[6-9]。单分子反应路径(E1和SN1)首先要对氨基进行质子化,而后脱除氨,当Cβ上H原子因活化而脱去时,发生E1消去反应;若亲核试剂进攻形成的碳正离子,则为SN1反应机理。双分子反应机理(E2和SN2)则始于季铵化合物的生成。当强碱存在于表面时,强碱会获取Cβ上的H并导致C—N键断裂,进而烯烃形成(即是E2反应机理);当表面存在亲核试剂时,亲核试剂攻击Cα并伴随着氨的脱除形成硫醇类化合物(即SN2机理)。相关的理论研究主要集中于含氮化合物在催化剂表面上的吸附,C—N键断裂的研究还很缺乏。因此,采用理论方法从原子水平阐述C—N键的断裂机理非常必要。过渡金属磷化物在HDN过程中的催化反应性能在近年来已得到了初步的研究和认可[10-12],其中磷化钼(MoP)具有很好的脱氮活性。
为认清杂环含氮化合物在磷化物表面上C—N键的断裂机理,本文利用DFT研究了哌啶在MoP(001)表面上的开环机理,并给出了相关反应的路径、结构及能量信息。由于含氮化合物HDN反应网络的复杂性,本文只研究HDN关键步骤——C—N键断裂的反应机理。本实验利用Materials Studio程序,基于DFT计算方法,对哌啶分子在MoP(001)表面上的吸附行为和C—N键断裂过程展开计算模拟。
2 实验过程
2.1 实验设备
实验仪器为Materials Studio软件,超算中心,计算机。
2.2 构建模型
构建模型环节可以分为如下几个步骤:①从软件自带的结构库中导入MoP的晶胞结构;②利用建模工具进行切面操作,建立超晶胞,设置真空层高度(需综合考虑模拟的真实性、计算精度和效率);③在MoP表面添加哌啶分子,需充分考虑分子在表面上各种可能的吸附位置,并通过结构优化确定最稳定的吸附位置。
如图1所示,Mo终端的MoP(001)表面采用5层厚度,每层含有9个原子(3×3)的超晶胞模型。晶胞之间采用1.3 nm的真空层,因此整个超晶胞高度是1.941 4 nm。模拟过程中,对MoP表面模型的上3层原子及哌啶分子进行无对称约束的结构优化,并固定底下两层原子于计算的体相点阵位置。
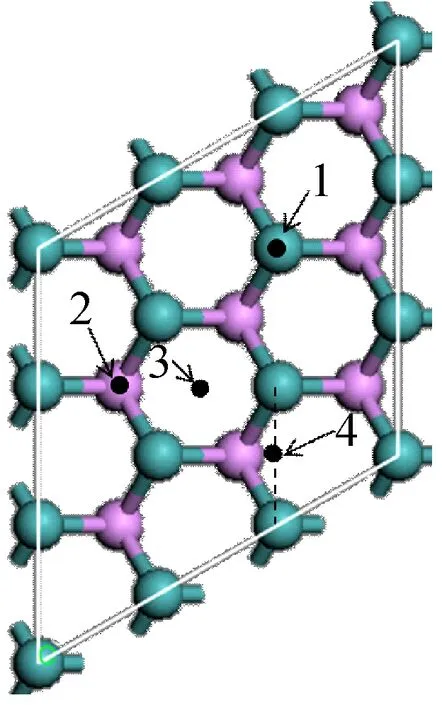
(a) 俯视图
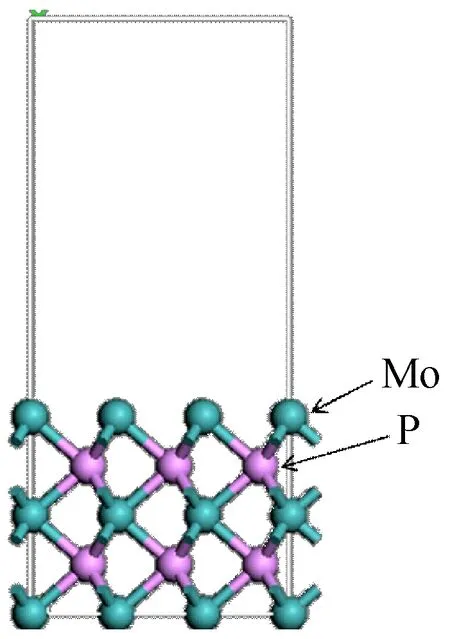
(b) 侧视图
2.3 计算设置
运用广义梯度近似(GGA)和PW91泛函相结合的方法(GGA-PW91)计算交换相关能[13-14]。采用双数值型基组和极化函数处理价电子波函数;对金属原子核内电子采用DFT半核芯赝势近似,对其他的原子,如氢、碳、氮和硫原子则采用全电子计算。积分计算采用Medium精度,其中能量、梯度和位移收敛标准分别为0.544 2 meV、1.088 eV/nm 和5×10-4nm。截断半径为0.5 nm,并用0.136 eV的轨道热占据来提高模拟精度。所有计算均采用自旋极化,每个表面只吸附一个吸附质分子,对应1/9的表面覆盖度。布里渊区内的积分采用2×2×1的Monkhorst-Packk点设置[15],格点间距大约为0.6 nm-1。采用p(4×4)的晶胞进行了测试计算,发现哌啶在最优吸附位的结合能与采用p(4×4)的晶胞确定的值之差小于0.02 eV,表明覆盖度效应可忽略。
本文采用的吸附能(Eads)计算公式:
Eads=Eadsorbate+EMoP(001)-Eadsorbate/MoP(001)
其中:Eadsorbate为气相哌啶分子的能量;EMoP(001)是MoP(001)清洁表面的能量;Eadsorbate/MoP(001)为adsorbate/MoP(001)吸附体系的总能量。吸附能为正值则表示该吸附为稳定的吸附。
过渡态(TS)的搜索是在与周期性结构计算相同理论等级上采用完全线性同步(LST)和二次同步变换(QST)方法[16]。首先最大化LST,然后在与反应路径共轭的方向取能量极小值,获得一个近似的TS,并用该TS进行QST的最大化,最后进行另一次共轭梯度的极小化。一直重复上述循环直到确定鞍点位置。通过频率分析所计算出的唯一虚频确认TS结构的正确性。
MoP(001)表面上一个基元反应的反应能(ΔE)和能垒(Ea)通过式计算:
ΔE=EFS/MoP(001)-EIS/MoP(001)
Ea=ETS/MoP(001)-EIS/MoP(001)
其中:EIS/MoP(001)、ETS/MoP(001)和EFS/MoP(001)分别对应于MoP(001)表面反应的初态(IS)、TS和反应末态(FS)的能量。
3 实验结果及讨论
3.1 哌啶的吸附特性
首先优化气相哌啶的分子结构,如图2所示,计算得到的键长为0.109 9~0.111 2 nm(C—H键)和0.102 0 nm(N—H键)。考虑到哌啶环的饱和特性,可以推测哌啶只能通过N的孤对电子与表面发生作用从而形成稳定吸附。计算结果证实哌啶只能通过N—Mo键吸附在Mo原子顶位。如图3所示,哌啶在MoP(001)表面上存在4种稳定的吸附构型,依次标记为hcp、fcc、top-1、top-2,分别表示哌啶环位于hcp、fcc和top位的上方。表1给出了这些构型对应的吸附能及重要的结构参数。吸附能分别为0.67 eV(hcp)、0.66 eV(fcc)、0.61 eV(top-1)和0.60 eV (top-2)。哌啶在MoP上较弱的吸附与其较长的N—Mo键(均大于0.230 nm)相吻合。此外,哌啶环中的相关键长并未因吸附而发生没有明显的改变,可认为C—N键并未受到活化作用。

图2 气相哌啶分子结构示意图
3.2 哌啶的C—N键断裂机理
实验表明,对于含氮杂环化合物的开环,没有证据表明其C—N键可以直接断裂。为确认该结论,首先研究了C—N键放热直接断裂反应,如图4所示。反应开始于hcp位吸附的哌啶,杂环开环后,相应的亚氨基结合在桥位而末端亚甲基占据邻近的hcp位。计算的能垒为2.36 eV,反应能为-0.68 eV。如此高的能垒表明杂环化合物中的C—N键确实难以直接发生断裂,这是因为哌啶与表面作用弱,C—N键的弱化程度较小。
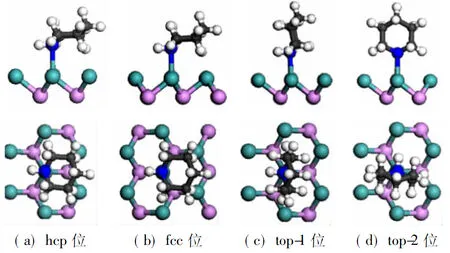
图3 哌啶在MoP(001)表面稳定吸附构型的侧视图和俯视图
表1 MoP(001)表面上哌啶的吸附位、吸附能及相关结构参数
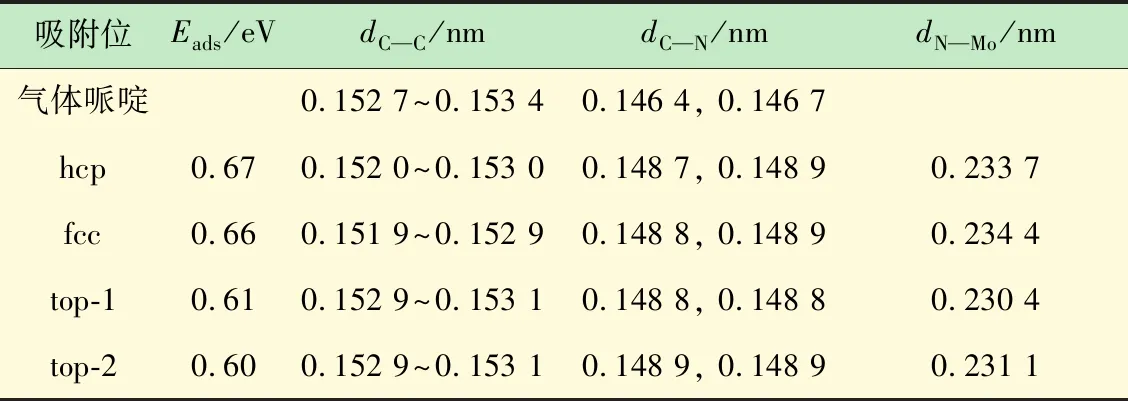
吸附位Eads/eVdC—C/nmdC—N/nmdN—Mo/nm气体哌啶0.152 7~0.153 40.146 4, 0.146 7hcp0.670.152 0~0.153 00.148 7, 0.148 90.233 7fcc0.660.151 9~0.152 90.148 8, 0.148 90.234 4top-10.610.152 9~0.153 10.148 8, 0.148 80.230 4top-20.600.152 9~0.153 10.148 9, 0.148 90.231 1

图4 哌啶在MoP(001)表面上的直接C—N键断裂反应
进一步考虑哌啶的C—N键断裂可能采取霍夫曼消去或是亲核取代反应机理。因此,哌啶必须通过N上的孤对电子与一个质子完成质子化的过程后,C—N键断裂才有可能发生。将MoP表面H原子作为哌啶质子化的质子来源,哌啶与MoP表面H原子的质子化反应见图5,反应始于hcp吸附的哌啶与哌啶环下方hcp位吸附的H,首先Mo—N键发生断裂,哌啶与氢原子互相靠近以使N的孤对电子与H结合,最终形成hcp位上方吸附的质子化哌啶,其哌啶环位于hcp位上方,且与表面成47°左右的夹角,N原子与最近Mo原子之间的距离为0.307 1 nm。在该质子化反应的过渡态(TS1-B)中,Mo和N原子间的距离为0.304 0 nm,Mo—N键已发生断裂,N与H之间的距离为0.145 3 nm。该反应的能垒是1.72 eV,反应能是1.47 eV。哌啶质子化完成后,可能的C—N键断裂路径有两条:消去(E1和E2)和亲核取代反应。在本计算中,MoP表面无酸碱性活性位和亲核试剂存在,C—N键只能通过E1消去机理断裂。质子化哌啶的一个C—N键在桥位作用下活化而发生断裂开环,形成5-aminopentyl-1(C5H12N)(见图5)。在该开环反应的过渡态(TS2-C)中,哌啶环已打开,C与N之间的距离为0.243 9 nm。该反应放热1.85 eV,对应的能垒较高,为1.73 eV,这是由于哌啶质子化后,其与表面的相互作用仍比较弱,由于开环反应涉及到环状碳链的展开,因而导致较高的反应能垒。由于E1消去过程(质子化+C—N键断裂)包含的反应能垒均比哌啶C—N键直接断裂能垒小很多,因此,哌啶通过E1消去而发生C—N键断裂开环是可行的。

图5 哌啶在MoP(001)表面上C—N键断裂的E1消去反应
4 结 语
本文对HDN反应过程进行计算实验设计,借助Materials Studio软件程序中的DMol3模块展开计算。依托超算中心平台,促进学生在掌握基本理论和模拟实验知识点的同时,也逐步熟悉了Linux系统上手操作能力。本实验进一步强化了学生的专业基础和科研素养。教学实践表明,学生更倾向于接受这种参与性较强,课程内容具有一定前沿性的研究型实验。
猜你喜欢
华东理工大学学报(自然科学版)(2022年2期)2022-04-29
健康体检与管理(2022年2期)2022-04-15
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
精神医学杂志(2021年2期)2021-06-23
中学生数理化(高中版.高考理化)(2019年10期)2019-11-08
中学化学(2019年1期)2019-06-29
山东化工(2019年1期)2019-01-24
中国药理学通报(2014年2期)2014-05-09
郑州大学学报(理学版)(2014年4期)2014-03-01
