
甲烷在钯基催化剂上脱氢的分子模拟
2020-07-02 02:43牟川淋蒲韵霜余洋邓淇铮唐子钰王林元邓洪波
石油与天然气化工 2020年3期
牟川淋 蒲韵霜 余洋 邓淇铮 唐子钰 王林元 邓洪波
1.西南石油大学化学化工学院 2.中国石油西南油气田公司成都天然气化工总厂
环境污染及能源短缺的问题逐渐引起了人们的高度重视,天然气具有储量丰富、价格低廉和实用性高等优点,是理想的能源之一。然而,由于天然气的不完全燃烧会导致NO2和CO等其他环境污染物的产生,解决这个问题最有效的方法之一,就是利用催化剂在低温下实现高效燃烧,即催化燃烧[1]。要实现CH4的高效燃烧,C-H键的活化是关键步骤[2-4]。因此,研究甲烷在催化剂作用下的脱氢过程,有利于找到提高甲烷燃烧效率的途径。Li等[5]在研究甲烷分子在铂原子(CH4-Pt)上的活化机制时,发现了一种新的低能量的脱氢途径。对于金属催化剂来说,单原子催化剂因其尺寸最小,能够最大限度地提高金属原子的使用效率,这对于贵金属催化剂尤为重要[6-9]。然而,二聚体同样作为小尺寸的催化剂,与单原子催化剂在催化活性上存在差异。Sun等[10]对甲烷在Fe原子以及二聚体Fe2的C-H键活化比较中发现,Fe2的催化活性高出Fe原子1~3倍。由于在二聚体催化剂中选择不同的两种金属,双金属之间存在相互影响,因此双金属二聚体催化剂能够进一步调节其催化性能。Wang等[11]比较了C-H在二聚体Pt2和PtNi催化剂上的活化能和热力学参数,发现两种催化剂上CH的形成是甲烷活化的RDS,并且铂二聚体Pt2的活性和抗炭沉积性能高于双金属催化剂PtNi。He等[12]采用DFT的B3LYP法对二聚体催化剂Ni2、NiCo和NiCu上甲烷脱氢反应进行了计算,得出NiCo具有较高的活性和抗炭沉积能力,在甲烷催化燃烧中表现出比其他两种催化剂更好的性能。钯基催化剂由于其较高的催化活性而被广泛关注。然而,对于钯基二聚体催化剂的甲烷脱氢反应过程,如过渡态和中间体,目前还不清楚。本研究采用密度泛函理论(DFT)中的M06L的方法,在M06L/6-311++G(d,p)+SDD//M06L/6-311G(d,p)+LANL2DZ基组水平对甲烷在二聚体Pd2和PdPt、PdNi催化剂上的脱氢过程进行了系统研究。基于催化剂Pd2、PdPt和PdNi上CHx(x=0~3)和H的共吸附,通过寻找反应过渡态来获得催化剂与甲烷反应的中间体,对比了甲烷与催化剂反应的几何优化结构、能垒、活化能和反应速率常数。为研究甲烷在钯基二聚体催化剂上脱氢的微观机理提供了参考,对甲烷燃烧催化剂的选择具有一定的指导作用。
1 计算部分
1.1 计算方法
采用Gaussian 09程序包中的DFT方法中的M06L,此密度泛函已被成功地应用于吸附和催化反应的研究[13-15],特别是对含有过渡金属体系的研究具有广泛的准确性[16-17]。在M06L/6-311G(d,p)+LANL2DZ基组水平上(C、H原子使用基组6-311G(d,p),金属原子Pd、Pt、Ni使用赝势基组LANL2DZ)对甲烷在二聚体催化剂Pd2、PdPt和PdNi上脱氢过程中的反应物、中间体、过渡态以及产物的构型进行了几何结构优化,并在相同水平上对中间体和过渡态进行了频率计算。对每个过渡态进行内禀反应坐标(IRC)计算,保证优化后的过渡态正确连接反应物和产物。为了得到更精确的结果,在M06L/6-311++G(d,p)+SDD基组水平上对反应路线上各驻点进行了单点能计算。根据过渡态理论,能量(Eb)由过渡态和初始状态之间的吉布斯自由能的差异呈现。反应能(Er)由最终状态和初始状态之间的焓差给出。
Eb=ΔG≠=GTS-GIS
(1)
Er=HFS-HIS
(2)
式中:GIS、GTS分别为吉布斯自由能初始状态和过渡状态;HIS、HFS分别为初始状态和最终状态的焓,kJ/mol。
在甲烷活化的DFT研究中,在Gaussian 09压力和温度保持恒定。由Eyring公式的热力学表达式结合Arrhenius公式,以及反应中的热力学、动力学参数,可以得到反应过程中的活化能(Ea),指前因子(A)和反应速率常数(k)[18]。
Ea=ΔH≠+nRT(n=1)
(3)
ΔG≠=-RTlnk≠=ΔH≠-TΔS≠
(4)
(5)
(6)
式中:T为反应温度,K;R为气体常数,J/(mol·K);ΔH≠为过渡态和初始态的焓差,kJ/mol;n为反应中涉及的分子数,n=1;ΔS≠为过渡态和初始状态之间的熵差,J/(mol·K);kB为玻耳兹曼常数,J/K;h为普朗克常数,J·s;k≠为热力学平衡常数。
1.2 反应机理
甲烷的脱氢过程由5个步骤组成[3,10-11,19],其反应途径如下所示:
CH4+M→CH3(s)+H(s)(M=Pd2,PdPt,PdNi)
(Ⅰ)
CH3(s)→CH2(s)+H(s)
(Ⅱ)
H(s)+H(s)→H2
(Ⅲ)
CH2(s)→CH(s)+H(s)
(Ⅳ)
CH(s)→C(s)+H(s)
(Ⅴ)
2 结果与讨论
2.1 甲烷脱氢的几何结构优化比较
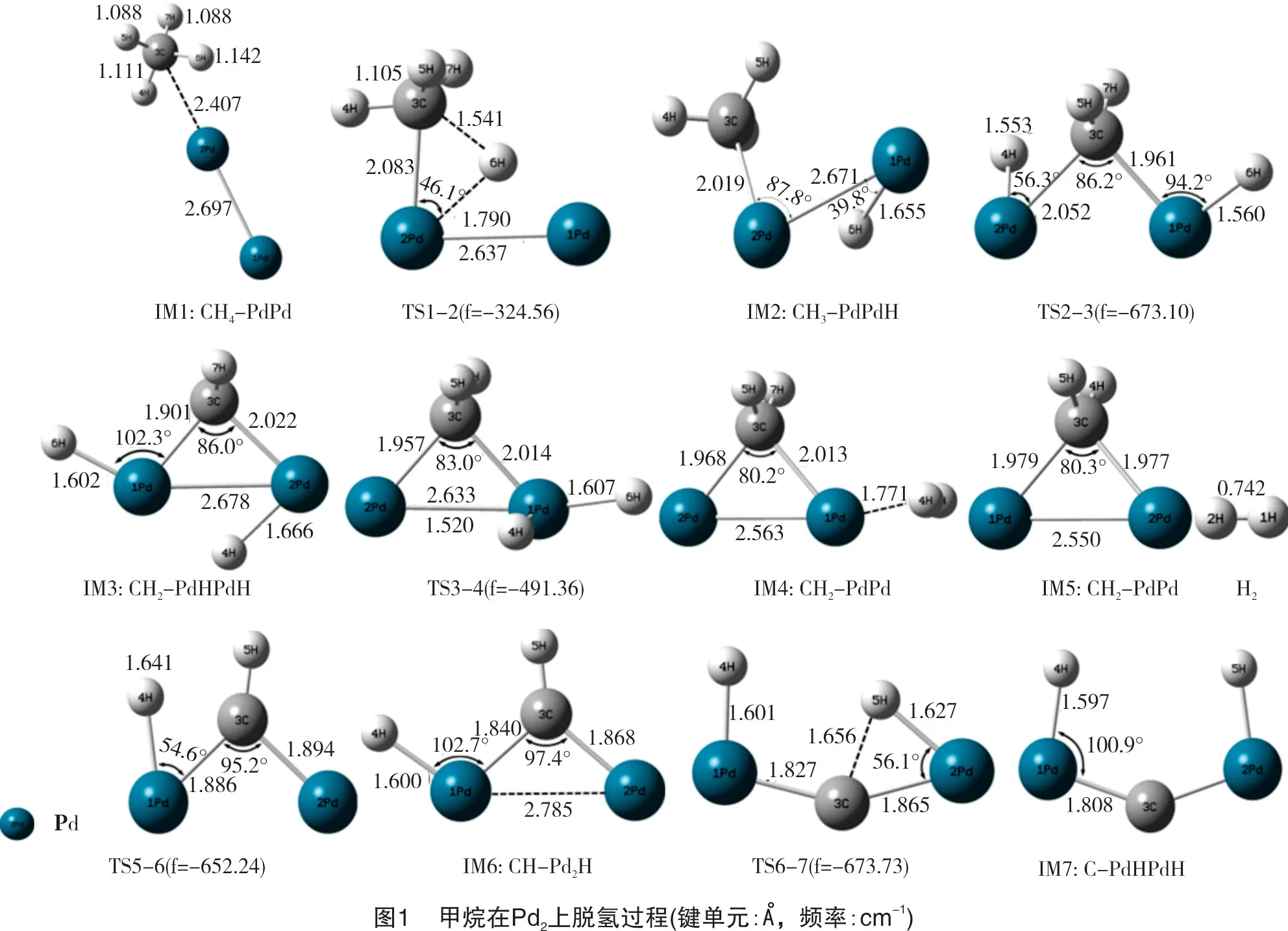
甲烷在钯二聚体Pd2上脱氢的反应物、中间体、产物、过渡态的优化结构和过渡态的虚频值如图1所示。在反应物IM1中,靠近催化剂的两个C-H键均有不同程度的伸长,最长的C-H键键长为1.142 Å(1 Å=0.1 nm,下同)。在过渡态TS1-2中,C-H键断裂,H原子向Pd原子靠近,CH3吸附在另一个Pd原子上,此时H-Pd键长为1.790 Å,C-Pd键长为2.083 Å。在IM2中,从CH4上脱下来的H原子与Pd原子形成单键,H-Pd键键长为1.655 Å。接着,第二个C-H键断裂,因为CH2是不饱和基团,所以其最稳定的吸附构型是与两个Pd原子结合形成桥位吸附。H原子被吸附到另一个Pd原子上,两个H-Pd键在IM3中有不同程度的延伸,键长分别为1.602 Å和1.666 Å。反应继续,H原子发生转移,与另外一个H原子结合形成H2。第三个C-H键断裂,H向催化剂中Pd原子上转移,并形成单键,键长为1.600 Å。第四个C-H键断裂,最后在产物IM7上,C与H共吸附在两个Pd原子上,C-Pd键长为1.806 Å,H-Pd键长为1.597 Å。每个优化反应物、产物和中间体的振动频率值均为正,表明其空间结构稳定。优化后的过渡态结构有且只有一个虚频,表明过渡态是合理的。IRC计算已确认过渡态正确连接相关的反应物和产物。
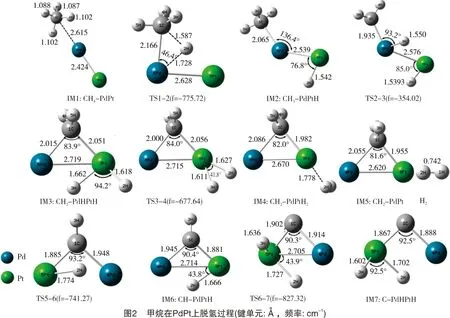
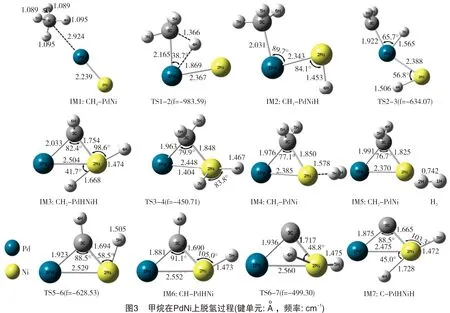
甲烷在催化剂Pd2与在PdPt和PdNi上的脱氢反应途径有一定的区别。甲烷在二聚体PdPt和PdNi上的脱氢反应途径分别如图2及图3所示。甲烷在双金属催化剂PdPt和PdNi的反应中,第一个C-H键断裂,H原子吸附到特征原子(Pt、Ni)上。在Pd2催化剂上,由于其本身无极性,两个H原子分别吸附在两个Pd原子上,而催化剂PdPt和PdNi两种金属原子之间存在极性,脱下来的两个H原子均吸附在特征原子上(Pt、Ni)。接着,特征原子上的两个H原子结合形成H2,说明甲烷在二聚体催化剂PdPt和PdNi上脱氢反应主要在特征原子(Pt、Ni)上进行,而Pd原子对反应起辅助作用。
2.2 甲烷脱氢的能垒比较
甲烷在二聚体Pd2上脱氢的势能分布如图4及表1所示。第一个C-H断裂(IM1→IM2),克服的能量为Eb=12.78 kJ/mol。第二个C-H键断裂(IM2→

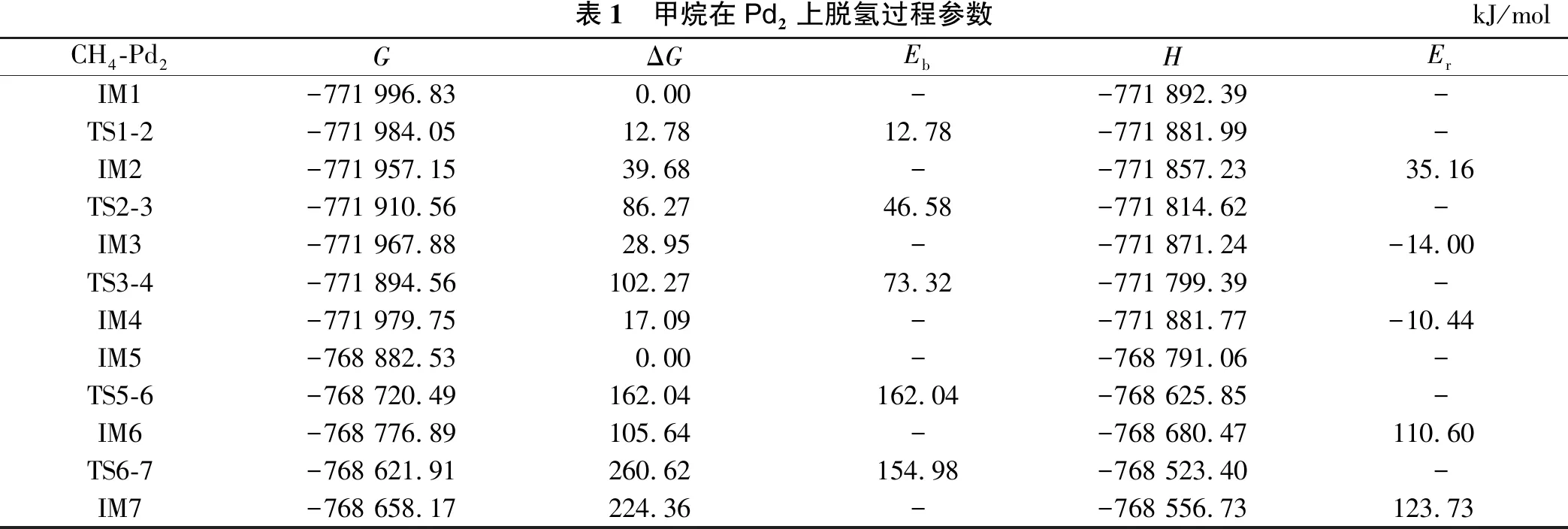
表1 甲烷在Pd2上脱氢过程参数kJ/molCH4-Pd2GΔGEbHErIM1-771 996.830.00--771 892.39-TS1-2-771 984.0512.7812.78-771 881.99-IM2-771 957.1539.68--771 857.2335.16TS2-3-771 910.5686.2746.58-771 814.62-IM3-771 967.8828.95--771 871.24-14.00TS3-4-771 894.56102.2773.32-771 799.39-IM4-771 979.7517.09--771 881.77-10.44IM5-768 882.530.00--768 791.06-TS5-6-768 720.49162.04162.04-768 625.85-IM6-768 776.89105.64--768 680.47110.60TS6-7-768 621.91260.62154.98-768 523.40-IM7-768 658.17224.36--768 556.73123.73
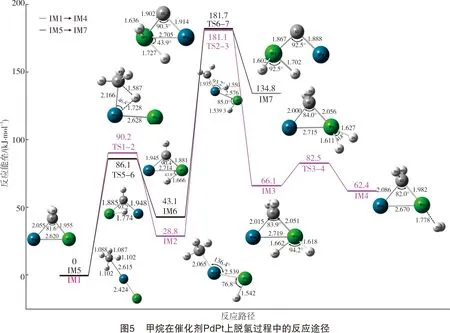
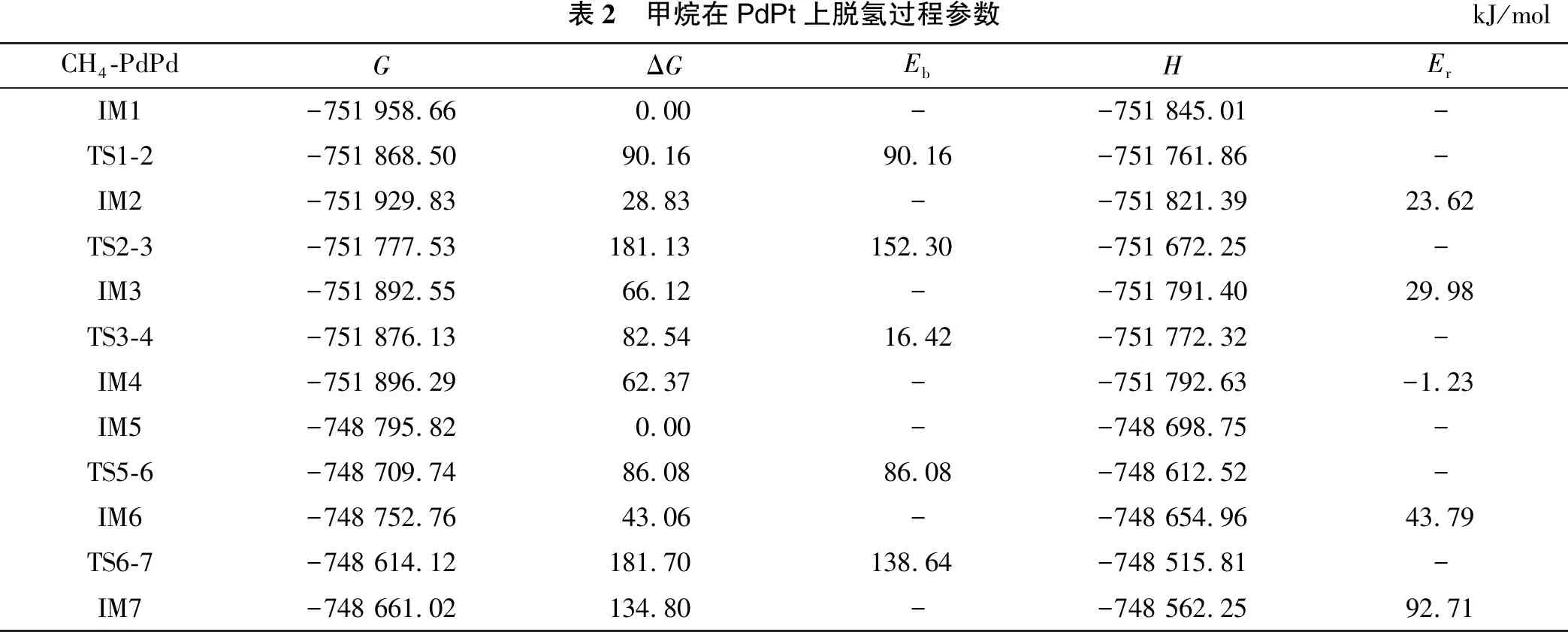
表2 甲烷在PdPt上脱氢过程参数kJ/molCH4-PdPdGΔGEb HErIM1-751 958.660.00--751 845.01-TS1-2-751 868.5090.1690.16-751 761.86-IM2-751 929.8328.83--751 821.3923.62TS2-3-751 777.53181.13152.30-751 672.25-IM3-751 892.5566.12--751 791.4029.98TS3-4-751 876.1382.5416.42-751 772.32-IM4-751 896.2962.37--751 792.63-1.23IM5-748 795.820.00--748 698.75-TS5-6-748 709.7486.0886.08-748 612.52-IM6-748 752.7643.06--748 654.9643.79TS6-7-748 614.12181.70138.64-748 515.81-IM7-748 661.02134.80--748 562.2592.71
IM3),Eb=46.58 kJ/mol。IM3→IM4是形成H2的过程。然后CH2吸收热量,第三个C-H键断裂(IM5→IM6),Eb=162.04 kJ/mol。第四个C-H键断裂(IM6→IM7),生成C和H共吸附在催化剂上,Eb=154.97 kJ/mol。在二聚体Pd2催化剂上,可以发现第三个C-H键裂解的能量势垒比其他3个C-H键裂解的能量势垒要高。因此,可以初步说明第三个C-H键裂解(IM5→IM6)可以作为CH4完全脱氢的速率控制步骤(RDS)。
甲烷在催化剂PdPt上脱氢的势能分布如图5及表2所示。在整个脱氢反应过程中,第二个C-H裂解的能量势垒最大(Eb=152.30 kJ/mol),因此初步确定第二个C-H键裂解为甲烷在二聚体催化剂PdPt脱氢过程中的RDS。
甲烷在催化剂PdNi上脱氢的能垒变化如图6及表3所示。在整个脱氢反应过程中,第二个C-H键断裂的能量势垒最大(Eb=178.10 kJ/mol),可初步确定第二个C-H键断裂为甲烷在二聚体催化剂PdNi上脱氢过程中的RDS。整个反应过程中,甲烷在二聚体Pd2上第三个C-H键断裂克服的能垒最大(Eb=162.04 kJ/mol),在催化剂PdPt、PdNi上第二个C-H键断裂克服的能垒最大,分别为152.30 kJ/mol、178.10 kJ/mol。因此,比较甲烷在3种二聚体催化剂上反应的最大能垒,可初步预测PdPt具有比Pd2和PdNi催化剂更高的催化活性。
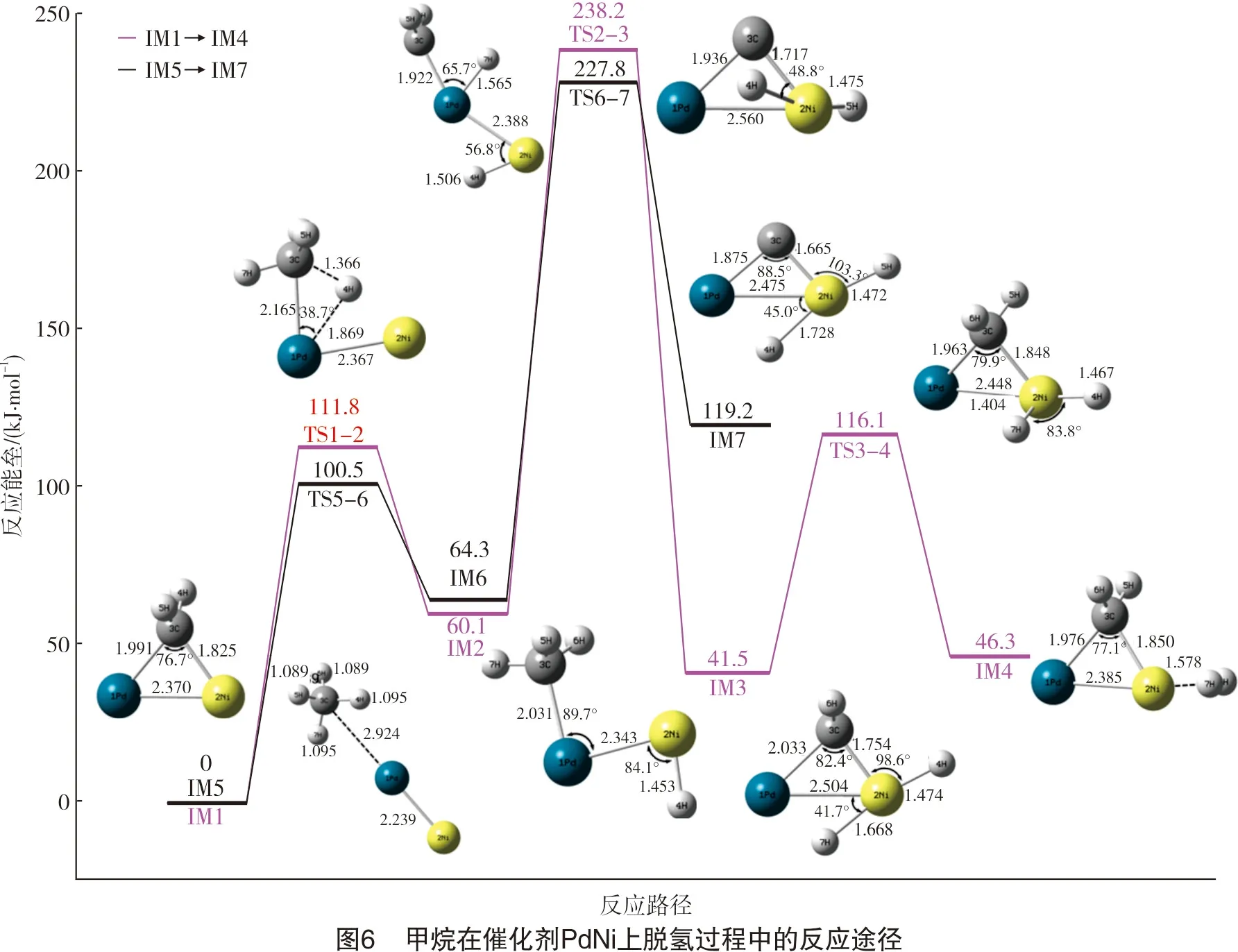
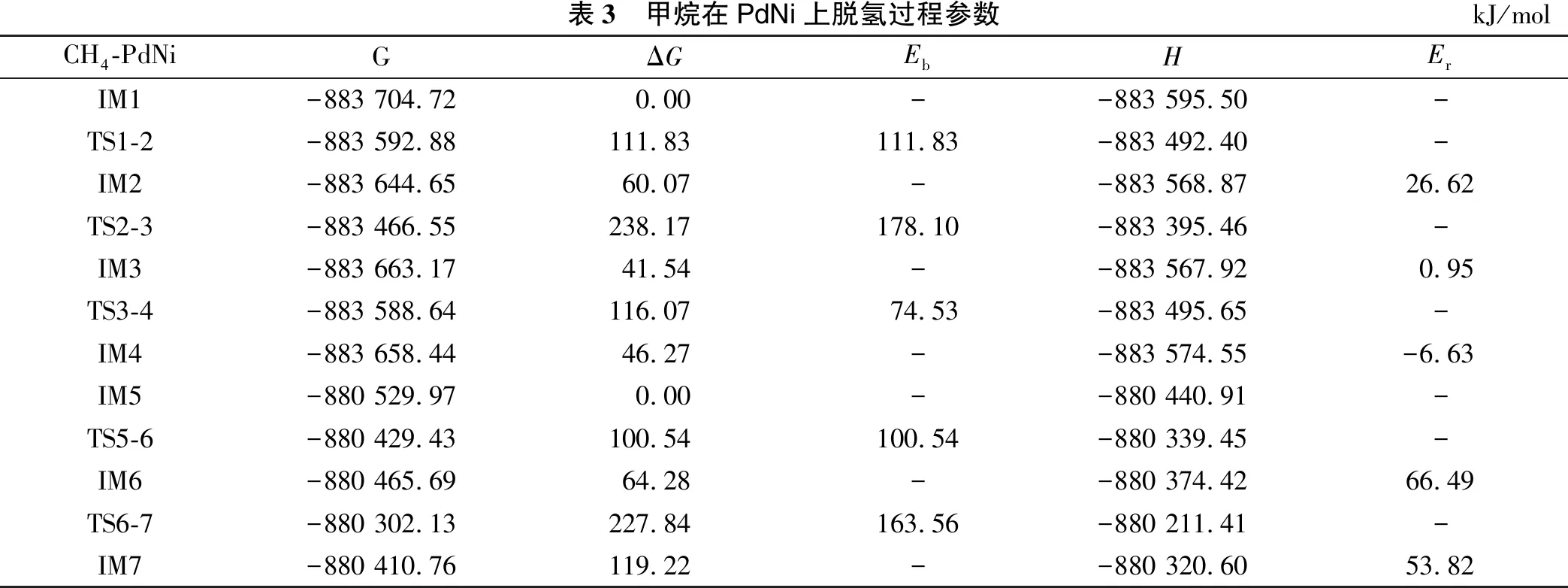
表3 甲烷在PdNi上脱氢过程参数kJ/molCH4-PdNiGΔGEbHErIM1-883 704.720.00--883 595.50-TS1-2-883 592.88111.83111.83-883 492.40-IM2-883 644.6560.07--883 568.8726.62TS2-3-883 466.55238.17178.10-883 395.46-IM3-883 663.1741.54--883 567.920.95TS3-4-883 588.64116.0774.53-883 495.65-IM4-883 658.4446.27--883 574.55-6.63IM5-880 529.970.00--880 440.91-TS5-6-880 429.43100.54100.54-880 339.45-IM6-880 465.6964.28--880 374.4266.49TS6-7-880 302.13227.84163.56-880 211.41-IM7-880 410.76119.22--880 320.6053.82
2.3 甲烷脱氢的热力学和动力学比较
甲烷在催化剂Pd2上的脱氢过程的热力学和Arrhenius参数如表4所示。在二聚体Pd2上CH4→CH3(IM1→IM2)反应活化能Ea=12.88 kJ/mol,反应速率常数k=3.57×1010s-1。反应CH3→CH2(IM2→IM3)发生,Ea=45.08 kJ/mol,k=4.24×104s-1。对于H(s)+H(s)→H2(IM3→IM4),Ea=74.32 kJ/mol,k=8.75×10-1s-1。CH2→CH(IM5→IM6)的反应活化能最大(Ea=167.69 kJ/mol),反应速率常数为最小值(k=2.46×10-16s-1)。反应CH→C(IM6→IM7)发生,Ea=159.54 kJ/mol,k=4.25×10-15s-1。这表明CH2→CH反应是最困难的,对所有基本步骤之间的反应总速率具有最大的影响,进一步说明CH2→CH是甲烷在二聚体Pd2反应的RDS。在之前的研究中,甲烷在Pd(111)上脱氢的RDS为CH3→CH2的形成[20],而在Pd(100)上脱氢的RDS为CH4→CH3的形成[21],这说明甲烷脱氢反应的RDS具有敏感性。
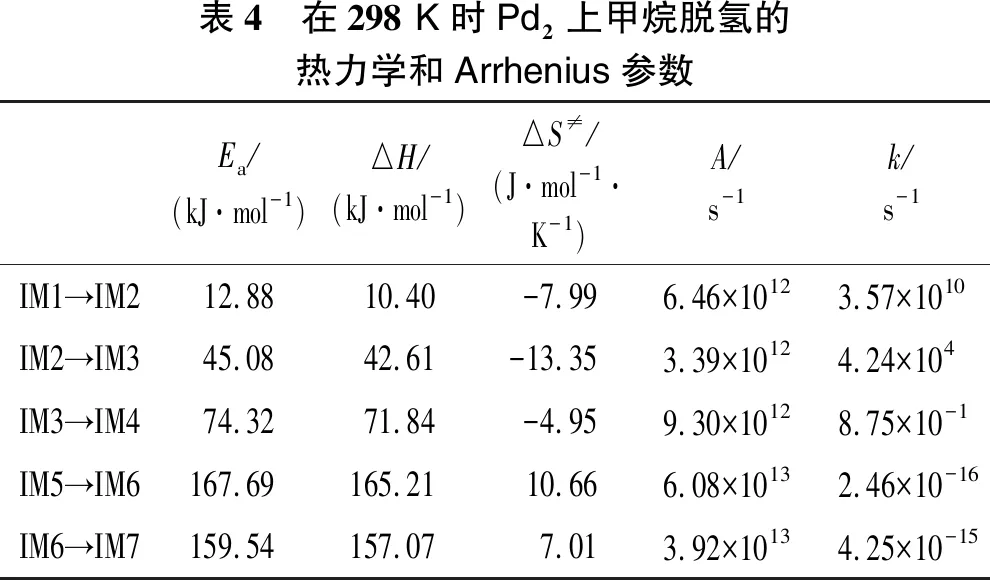
表4 在298 K时Pd2上甲烷脱氢的热力学和Arrhenius参数Ea/(kJ·mol-1)△H/(kJ·mol-1)△S≠/(J·mol-1·K-1)A/s-1k/s-1IM1→IM212.8810.40-7.996.46×10123.57×1010IM2→IM345.0842.61-13.353.39×10124.24×104IM3→IM474.3271.84-4.959.30×10128.75×10-1IM5→IM6167.69165.2110.666.08×10132.46×10-16IM6→IM7159.54157.077.013.92×10134.25×10-15
甲烷在催化剂PdPt、PdNi上的脱氢过程的热力学和Arrhenius参数分别如表5和表6所示。在双金属PdPt中,IM2→IM3的反应活化能(Ea=151.61 kJ/mol)是最大的,而反应速率常数(k=1.25×10-14s-1)是整个反应中最小的。同样,甲烷在双金属PdNi中,IM2→IM3反应活化能也最大(Ea=175.89 kJ/mol),反应速率常数最小(k=3.76×10-19s-1)。因此,说明反应CH3→CH2是催化剂PdPt、PdNi上甲烷活化的RDS。
在整个脱氢反应中,CH2→CH是二聚体Pd2的RDS,而CH3→CH2是催化剂 PdPt和PdNi的RDS,说明甲烷脱氢反应的速率控制步骤对催化剂具有敏感性。在3种催化剂的RDS反应上,催化剂PdPt的活化能最小(Ea=151.61 kJ/mol),PdNi活化能最大(Ea=175.89 kJ/mol),催化剂PdPt上脱氢的反应速率常数(k=1.25×10-14s-1)大于Pd2对甲烷脱氢的反应速率常数(k=2.46×10-16s-1),PdNi的反应速率常数是最小的(k=3.76×10-19s-1),这表明催化剂活性由大到小为PdPt>Pd2>PdNi。反应CH(s)→C(s)+H(s)可评价炭沉积的作用,活化能越大,反应速率常数越小,说明越能抗炭沉积。在PdNi催化剂上活化能最大(Ea=165.49 kJ/mol),反应速率最小(k=1.33×10-16s-1),说明PdNi催化剂更能抗炭沉积。
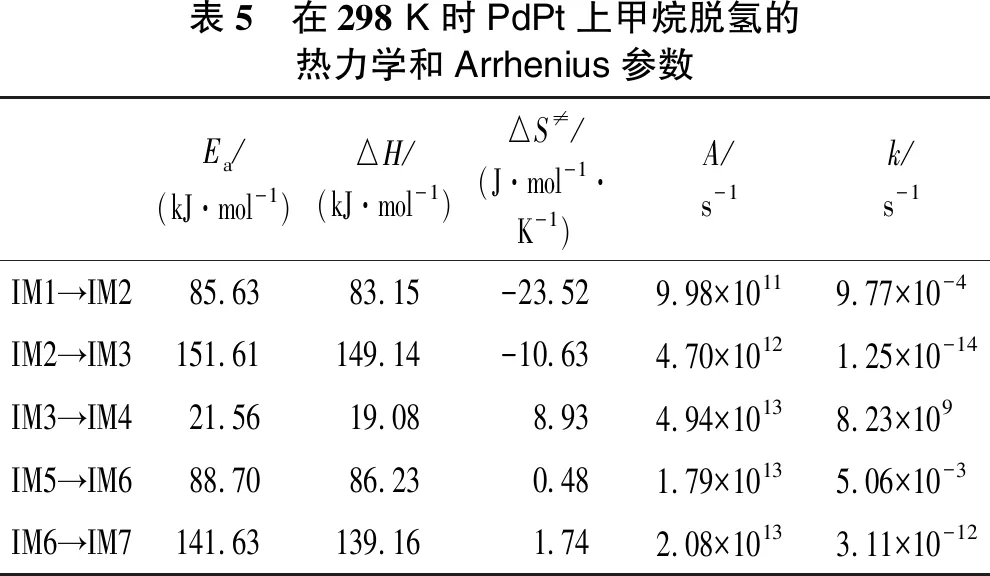
表5 在298 K时PdPt上甲烷脱氢的热力学和Arrhenius参数Ea/(kJ·mol-1)△H/(kJ·mol-1)△S≠/(J·mol-1·K-1)A/s-1k/s-1IM1→IM285.6383.15-23.529.98×10119.77×10-4IM2→IM3151.61149.14-10.634.70×10121.25×10-14IM3→IM421.5619.088.934.94×10138.23×109IM5→IM688.7086.230.481.79×10135.06×10-3IM6→IM7141.63139.161.742.08×10133.11×10-12
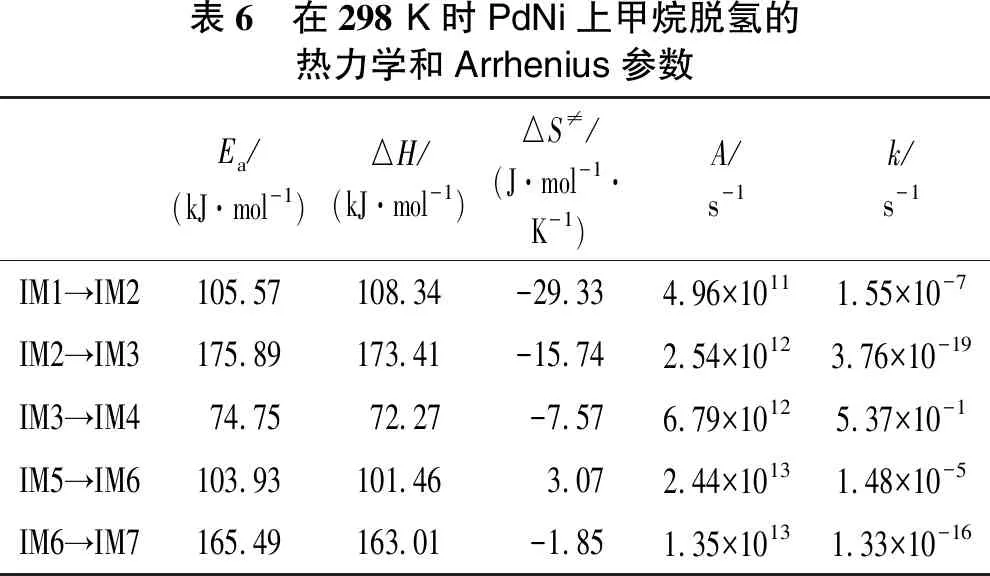
表6 在298 K时PdNi上甲烷脱氢的热力学和Arrhenius参数Ea/(kJ·mol-1)△H/(kJ·mol-1)△S≠/(J·mol-1·K-1)A/s-1k/s-1IM1→IM2105.57108.34-29.334.96×10111.55×10-7IM2→IM3175.89173.41-15.742.54×10123.76×10-19IM3→IM474.7572.27-7.576.79×10125.37×10-1IM5→IM6103.93101.463.072.44×10131.48×10-5IM6→IM7165.49163.01-1.851.35×10131.33×10-16
3 结论
本研究采用密度泛函理论(DFT)中的M06L的方法,在M06L/6-311++G(d,p)+SDD//M06L/6-311G(d,p)+LANL2DZ基组水平上,对甲烷在二聚体Pd2和PdPt、PdNi催化剂上的脱氢过程进行了系统研究。研究结果如下:
(1) 反应CH2→CH是甲烷在催化剂Pd2上反应的RDS,而反应CH3→CH2是甲烷在催化剂PdPt和PdNi上反应的RDS。
(2) 甲烷在3种催化剂反应的RDS上,其活化能从大到小依次为:PdNi(Ea=175.89 kJ/mol)> Pd2(Ea=167.69 kJ/mol)>PdPt(Ea=151.61 kJ/mol),反应速率常数从大到小依次为:PdPt(k=1.25×10-14s-1)>Pd2(k=2.46×10-16s-1)>PdNi(k=3.76×10-19s-1)。从催化活性这方面来看,3种催化剂中PdPt的催化活性最高。
(3) 在反应CH→C中,甲烷在3种催化剂上反应的活化能从大到小依次为:PdNi(Ea=165.49 kJ/mol)>Pd2(Ea=159.54 kJ/mol)>PdPt(Ea=141.63 kJ/mol),反应速率常数从大到小依次为:PdPt(k=3.11×10-12s-1)>Pd2(k=4.25×10-15s-1)>PdNi(k=1.33×10-16s-1)。从抗积炭性能来看,PdNi抗炭沉积性能良好。
(4) 从催化活性来看,催化剂PdPt反应速率常数最大,催化活性最好,适用于要求催化效率更高的项目。而催化剂PdNi催化效果虽不如Pd2、PdPt,但PdNi抗炭沉积性能良好且成本低,适合大型工业化项目。
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
中学生数理化(高中版.高考理化)(2021年5期)2021-07-16
装备维修技术(2020年5期)2020-11-20
矿产综合利用(2020年1期)2020-07-24
电脑知识与技术(2018年3期)2018-03-21
分析化学(2018年1期)2018-01-18
分析化学(2017年9期)2017-10-16
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
电子制作(2016年19期)2016-08-24
