
双氟磺草胺的合成新工艺
2020-12-26 04:44高士光蔡旭阳
现代农药 2020年6期
高士光,蔡旭阳,薛 欢,陶 涛
(江苏好收成韦恩农化股份有限公司,江苏启东 226200)
双氟磺草胺(Florasulam)是由美国陶氏农业科学公司开发的三唑并嘧啶磺酰胺类除草剂,是继磺草唑胺、唑嘧磺草胺、氯酯磺草胺与双氯磺草胺之后于20世纪90年代中期成功开发的第5个三唑并嘧啶磺酰胺类除草剂新品种[1]。其化学结构式见图1。
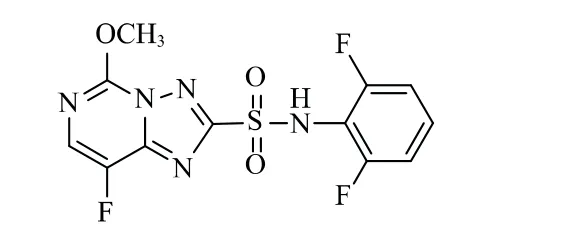
图1 双氟磺草胺化学结构式
双氟磺草胺是典型的乙酰乳酸合成酶(ALS)抑制剂,属于内吸传导型除草剂,可以传导至杂草全株,因而杀草彻底。其具有低毒高效、对环境友好、持效性好等特点,尤其是在低温下药效稳定,即使是在2℃时仍能保证药效稳定。双氟磺草胺能够用于多种作物,尤其是对麦类作物与草坪具有高度选择性,使用有效剂量仅为3~10 g/hm2[2-3]。2019年全球双氟磺草胺市场总值达到了17亿元,预计至2026年可以增长到35亿元,年复合增长率为10.5%,市场前景广阔[4]。
1 合成路线
国内有关双氟磺草胺合成工艺研究报道较少,通过对文献资料的调研和总结,目前其合成方法主要有2种。其合成路线1[5]见图2。该合成路线以5-氟尿嘧啶为起始原料,经氯化反应、醚化反应、肼基化反应、环合反应、转位反应、偶联反应、氯氧化反应和磺酰胺缩合反应,最终得到双氟磺草胺。
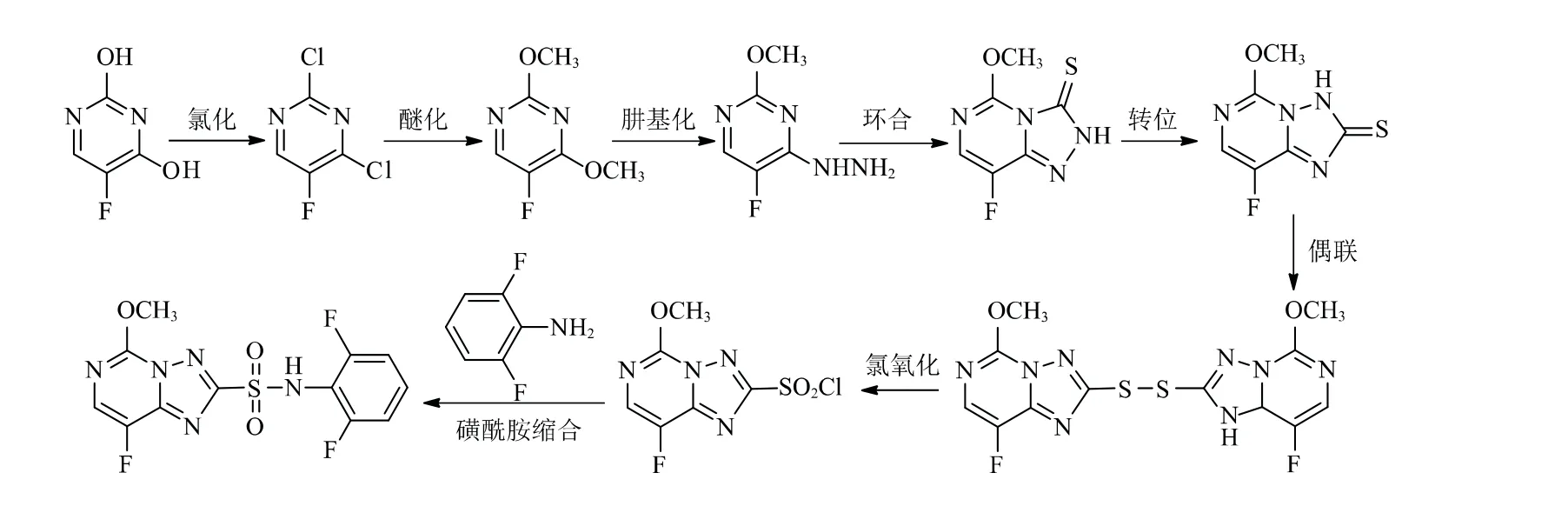
图2 双氟磺草胺合成路线1
双氟磺草胺合成路线2[6-9]见图3。该合成路线以2,4-二氯-5-氟嘧啶为起始原料,经醚化反应、肼基化反应、环合反应、苄硫醚化反应、异构化反应、氯氧化反应和磺酰胺缩合反应,得到双氟磺草胺。
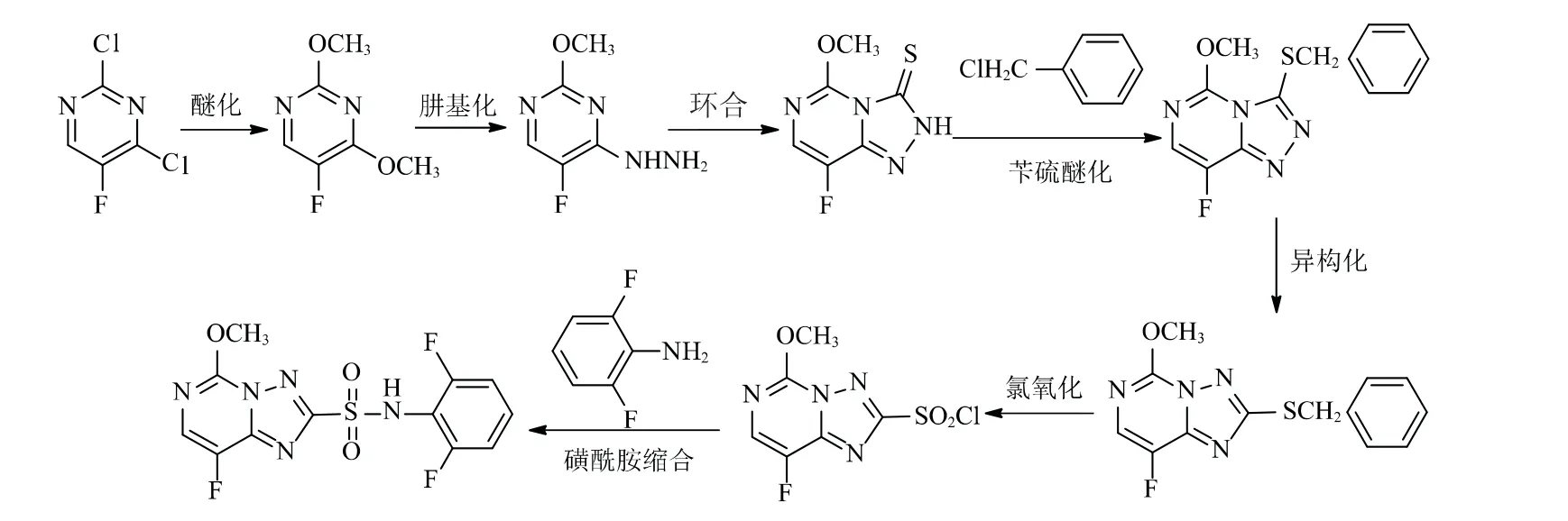
图3 双氟磺草胺合成路线2
以上2条合成路线,都需要用到化合物2,4-二氯-5-氟嘧啶,但现阶段制备该化合物的原料为5-氟尿嘧啶。由于厂家停限产较多,环保压力过大,开工率低,造成5-氟尿嘧啶市场供应紧张,报价高位,成本过高。
针对双氟磺草胺合成工艺中最后1步磺酰胺缩合反应,通过文献分析和总结发现,路线1和路线2参考了专利WO9937650A1[10]公开的制备方法,在1,2-丙二醇和萘催化剂的作用下,使用至少3倍量的2,6-二氟苯胺制备双氟磺草胺,而多余的2,6-二氟苯胺无法回收利用,从而导致生产成本较高,不适合工业化生产。专利CN1216040A[11]公开了一种双氟磺草胺的制备方法,该方法是先将2,6-二氟苯胺制成N-(2,6-二氟苯基)-S,S-二甲基硫亚胺,然后再用其催化(催化量为7.8%)制备双氟磺草胺。其不足在于转化率较低,只有70%,故而不适合工业化生产。同时,专利CN103509027A[12]公开了一种双氟磺草胺的制备方法,该方法虽然收率较高,而且引入价格低廉的三乙胺替代过量的2,6-二氟苯胺以降低生产成本。但是,经过大量实验论证发现,该方法重复性较差,对物料的配比以及反应温度和反应时间要求较高,容易产生杂质,因此不适合工业化生产。
笔者通过查阅相关文献资料[13-14]并结合上述合成方法[5-12],对双氟磺草胺合成工艺进行优化,用价格相对低廉的2-甲氧基-5-氟尿嘧啶为起始原料,制备得到关键中间体2-氯磺酰基-8-氟-5-甲氧基[1,2,4]三唑[1,5-c]嘧啶,然后再与NCS、甲硫醚、3-甲基吡啶和2,6-二氟苯胺通过磺酰胺缩合反应制备双氟磺草胺,能够获得较高的反应收率和产物纯度。优化后的工艺,合成条件温和,生产成本较低,重复性好,适合工业化生产。改进后的双氟磺草胺合成路线3见图4。
2 实验部分
2.1 仪器与试剂
(1)主要仪器。AVANCE Ⅲ400M核磁共振波谱仪,瑞士布鲁克公司;岛津LC-20A高效液相色谱仪(HPLC),日本岛津公司;旋转蒸发仪、SHZ-CD型循环水式多用真空泵、DFY-5L/40低温恒温反应浴、DHG-9030A电热恒温鼓风干燥箱,巩义市予华仪器有限责任公司。
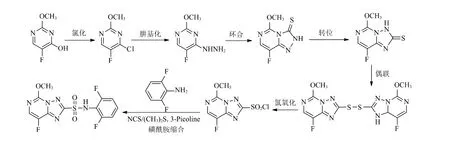
图4 双氟磺草胺合成路线3
(2)主要试剂。中间体2-氯磺酰基-8-氟-5-甲氧基[1,2,4]三唑[1,5-c]嘧啶(按照文献自制);2,6-二氟苯胺(99%,工业级),扬州天辰精细化工有限公司;3-甲基吡啶(99%,工业级),潍坊绿霸化工有限公司;N-氯代琥珀酰亚胺(98%,工业级),山东西亚化工科技有限公司;甲硫醚(99%,工业级),上海诺泰化工有限公司;其他试剂均为市售化学纯或分析纯级别。
2.2 实验步骤
2.2.1 2-甲氧基4-氯-5-氟嘧啶的合成
在500 mL带温度计、冷凝管和机械搅拌的四口烧瓶中,先将2-甲氧基-5-氟尿嘧啶28.8 g(0.2 mol/L)、三氯氧磷46 g(0.3 mol/L)及甲苯300 mL搅拌,滴加三乙胺30.3 g(0.3 mol/L)放热,控制温度不超过80℃。滴完升温至100~110℃,反应4 h后冷却至30℃,再将反应液滴入300 g冰水中,控温不超过10℃。滴完,搅拌30 min,静置分层,有机层用水洗1次,减压蒸馏去除溶剂后得到残余物2-甲氧基-4-氯-5-氟嘧啶共计30 g,粗品收率92.3%,纯度98%。
2.2.2 2-甲氧基-4-肼基-5-氟嘧啶的合成
在500 mL带温度计、冷凝管和机械搅拌的四口烧瓶中,先加入2-甲氧基4-氯-5-氟嘧啶30 g(0.185 mol/L),甲醇200 g,碳酸钠19.6 g(0.185 mol/L),水20 g,降温至0℃,滴加水合肼30 g(0.555 mol/L),滴完后,慢慢恢复至室温,然后加热回流反应4.5 h。反应结束后,冷却至0℃,析出固体,抽滤,甲醇(冷藏)100 g洗涤滤饼,干燥后得2-甲氧基-4-肼基-5-氟嘧啶25.7 g,收率88%,纯度大于97.5%。
2.2.3 8-氟-5-甲氧基-1,2,4-三唑[4,3-c]-3(2H)-硫酮的合成
在500 mL带温度计、冷凝管和机械搅拌的四口烧瓶中,先加入2-甲氧基-4-肼基-5-氟嘧啶25.7 g(0.16 mol/L),甲醇80 g,三乙胺24.3 g(0.24 mol/L),以及二硫化碳18.3 g(0.24 mol/L)。氮气置换后,开动搅拌,将反应液的温度降至15℃左右,将双氧水23.7 g(27.5%,0.192 mol/L)滴入上述反应体系中,整个滴加过程持续1 h左右,期间温度维持在15℃左右,滴完后反应10 h。反应结束后,过滤除掉生成的硫,滤液用10%盐酸溶液酸化至pH=3,过滤,真空干燥得到固体,共计30 g,收率93.7%,纯度大于95%。
2.2.4 8-氟-5-甲氧基-1,2,4-三唑[1,5-c]-2(3H)-硫酮的合成
在500 mL带温度计、冷凝管和机械搅拌的四口烧瓶中,先加入甲醇100 g,氢氧化钠18 g(0.45 mol/L)。溶清后,将8-氟-5-甲氧基-1,2,4-三唑[4,3-c]-3(2H)-硫酮30 g(0.15 mol/L)分批加入,反应3~4 h。反应结束后,降温至小于10℃,用10%盐酸溶液酸化至pH=3,过滤,水洗,得固体25.8 g,收率86%,纯度大于95%。
2.2.5 2,2′-二联硫(8-氟-5-甲氧基-1,2,4-三唑[1,5-c]嘧啶)的合成
在500 mL带温度计、冷凝管和机械搅拌的四口烧瓶中,加入8-氟-5-甲氧基-1,2,4-三唑[1,5-c]-2(3H)-硫酮25.8 g(0.129 mol/L),甲醇120 g,开动搅拌,将反应液降温至20℃以下,滴加双氧水31.8 g(27.5%,0.258 mol/L)。随着双氧水的滴加,反应体系的温度略有升高,最高升至38℃,维持温度在35~40℃,反应4~5 h,HPLC跟踪分析至底物含量小于0.1%,停止反应,降温抽滤,固体用少量甲醇漂洗,真空干燥,得产品22.6 g,收率88%,纯度大于95%。
2.2.6 2-氯磺酰基-8-氟-5-甲氧基[1,2,4]三唑[1,5-c]嘧啶的合成
在500 mL带温度计、冷凝管和机械搅拌的四口烧瓶中的反应器中,加入2,2′-二联硫(8-氟-5-甲氧基-1,2,4-三唑[1,5-c]嘧啶)22.6 g(0.056 mol/L)、二氯甲烷200 mL和水200 mL搅拌,用冰水浴冷却。在0~5℃,慢慢通入氯气32.2 g(0.448 mol/L),保持内温不超过10℃。通完后在0~10℃,继续搅拌反应30~60 min,分出有机相,用无水硫酸镁干燥,过滤,滤液脱溶后,得26.6 g黄白色状固体2-氯磺酰基-8-氟-5-甲氧基[1,2,4]三唑[1,5-c]嘧啶,收率90%,纯度大于95.6%。
2.2.7 双氟磺草胺的合成
在氮气保护下,向500 mL的四口反应瓶中加入乙腈100 mL和2,6-二氟苯胺12.9 g(0.1 mol/L),搅拌降温至-20℃,滴加甲硫醚3.1 g(0.05 mol/L),滴完搅拌30 min,再缓慢滴加26.67 g的N-氯代琥珀酰亚胺的乙腈溶液(N-氯代琥珀酰亚胺6.67 g,0.05 mol/L)。滴完继续反应1~2 h,然后缓慢滴加3-甲基吡啶27.9 g(0.3 mol/L),搅拌1 h后,再将56.6 g的2-氯磺酰基-8-氟-5-甲氧基[1,2,4]三唑[1,5-c]嘧啶的乙腈溶液(含2-氯磺酰基-8-氟-5-甲氧基[1,2,4]三唑[1,5-c]嘧啶26.6 g,0.1 mol/L)缓慢滴加到上述反应液中,1 h内滴完。然后,先于室温搅拌1 h,再升温至40℃,反应3~4 h,HPLC跟踪至2,6-二氟苯胺含量小于0.5%,结束反应。
反应结束后,将反应液温度降至0℃以下,滴入稀酸水250 g(质量分数为35%的盐酸41.7 g),酸化至pH小于2。析出大量固体,搅拌1 h,抽滤,滤饼水洗,少量甲醇淋洗,烘干,得到白色粉末状双氟磺草胺33.8 g,收率为94.2%,纯度为98.5%,熔点220~223℃(文献值为193.5~230.5℃),其高效液相色谱图见图5。
1H NMR (400 MHz, DMSO-d6)δ:4.226(s,3H,OCH3)、6.723 ~7.294(m,3H,苯环氢)、7.963(d,J=2.0 Hz,1H,NCH=CF)、10.664(s,1H,NHSO2)。

图5 双氟磺草胺高效液相色谱图
3 结果与讨论
3.1 2-甲氧基-4-氯-5-氟嘧啶的合成影响因素
该氯化反应的主要影响因素为2-甲氧基-5-氟尿嘧啶、三氯氧磷和三乙胺的摩尔比,在其他条件不变的情况下,考察得出最优的反应摩尔比,结果见表1。
由表1可知,2-甲氧基-5-氟尿嘧啶、三氯氧磷和三乙胺的摩尔比为1∶1.5∶1.5时最为合适,收率达92.3%。三氯氧磷用量少,反应收率偏低;三氯氧磷用量多,反应收率基本不变,成本增加。三乙胺作为缚酸剂的同时还能起到一定的催化作用,其用量和三氯氧磷同步,反应效果最好。
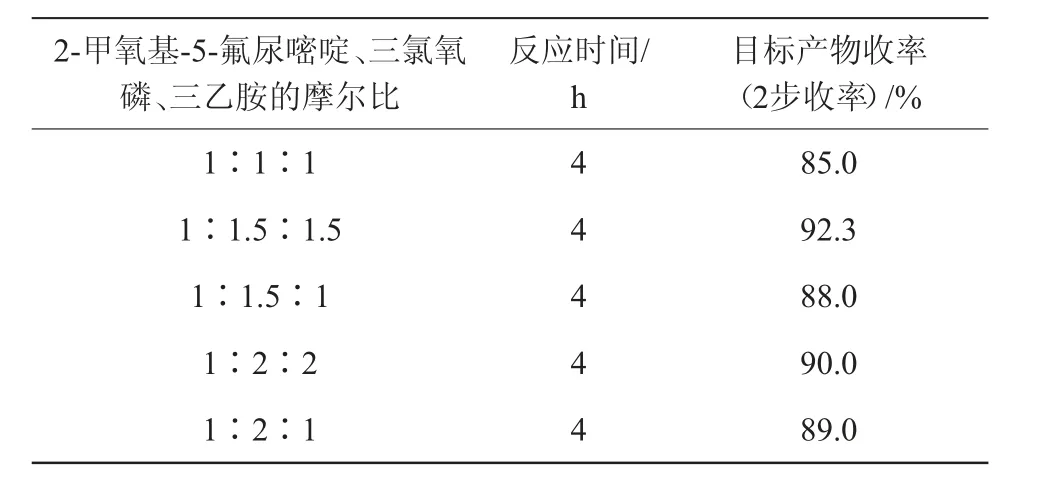
表1 2-甲氧基-5-氟尿嘧啶、三氯氧磷和三乙胺的摩尔比对目标产物收率的影响
3.2 2-甲氧基-4-肼基-5-氟嘧啶的合成影响因素
该肼基化反应的主要影响因素是2-甲氧基-4-氟-5-氟嘧啶与水合肼的摩尔比和反应时间。如果水合肼的用量过少,会导致反应时间过长,副产物增多,结果见表2。

表2 2-甲氧基-4-氟-5-氟嘧啶与水合肼的摩尔比和反应时间对目标产物收率的影响
由表2可知,2-甲氧基-4-氟-5-氟嘧啶与水合肼摩尔比为1∶3,反应时间为4.5 h时,最为合适,收率达95%。当水合肼的用量少时(小于3倍量),反应时间延长,转化率低;水合肼的用量多时(大于3倍量),转化率并没有太大的提高,但成本增加。
3.3 8-氟-5-甲氧基-1,2,4-三唑[4,3-c]-3(2H)-硫酮的合成影响因素
该环合反应的主要影响因素是2-甲氧基-4-肼基-5-氟嘧啶与二硫化碳的摩尔比及反应时间。在其他条件不变的情况下,考察得出了最优的反应摩尔比,结果见表3。
由表3可知,2-甲氧基-4-肼基-5-氟嘧啶与二硫化碳摩尔比为1∶1.5,反应时间为10 h时,最为合适,收率达93.7%。增加二硫化碳的比例,同时延长反应时间,收率没有增加。
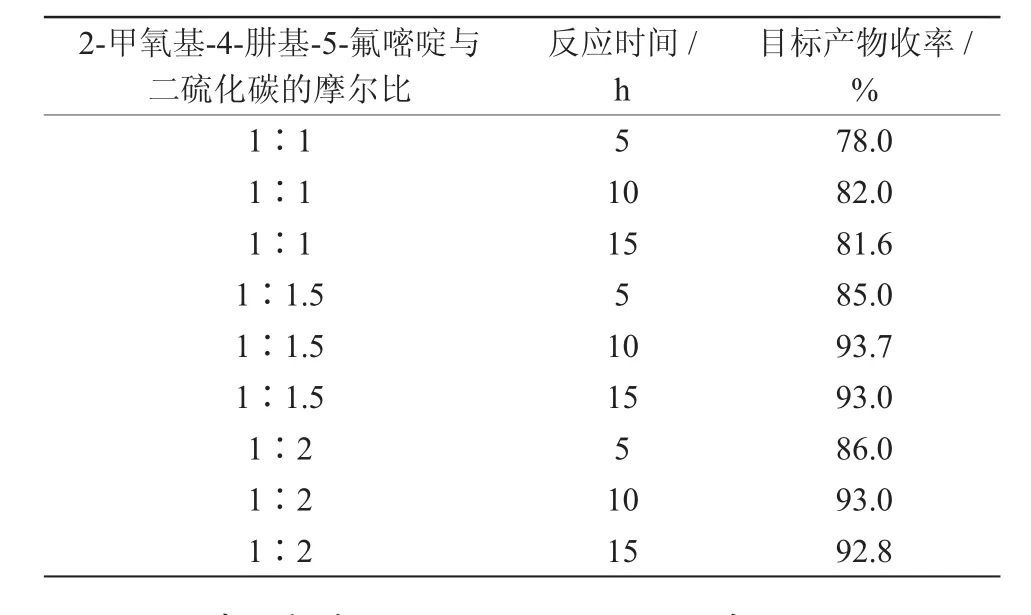
表3 2-甲氧基-4-肼基-5-氟嘧啶与二硫化碳的摩尔比和反应时间对目标产物收率的影响
3.4 双氟磺草胺的合成影响因素
该酰胺化反应的主要影响因素是2,6-二氟苯胺与甲硫醚和N-氯代琥珀酰亚胺的摩尔比及反应温度。因此,笔者重点考察了这2个因素,得出一个较优合成工艺条件,结果见表4和表5。
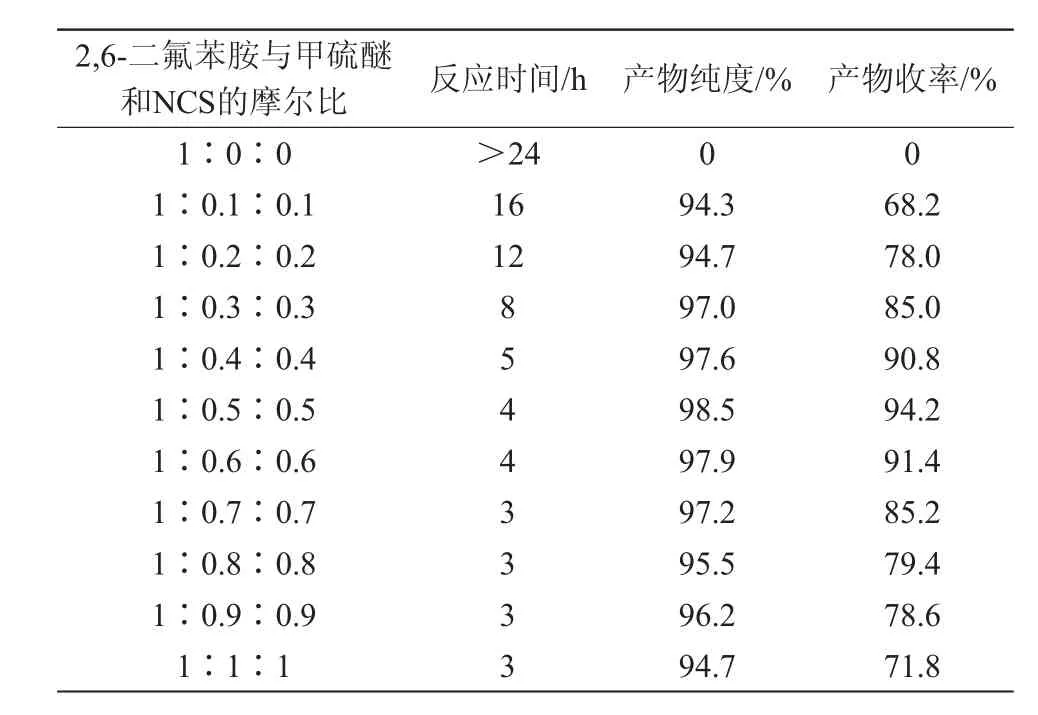
表4 2,6-二氟苯胺与甲硫醚、NCS 的摩尔比对目标产物收率的影响
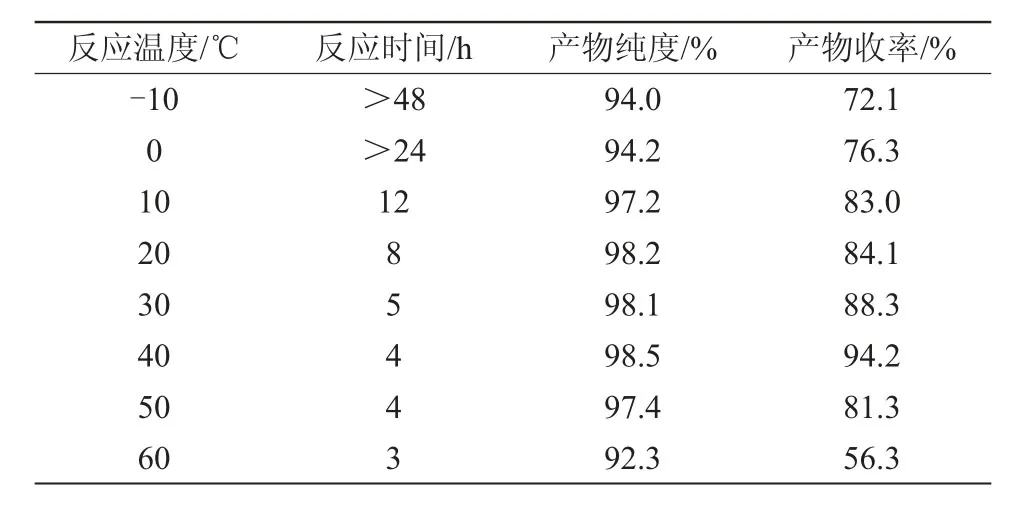
表5 反应温度对目标产物收率的影响
3.4.1 2,6-二氟苯胺与甲硫醚、NCS的摩尔比对目标产物收率的影响
由表4可知,在其他反应条件不变的情况下,2,6-二氟苯胺与甲硫醚和NCS的摩尔比为1∶0.5∶0.5,反应时间4 h时,反应收率高,目标产物纯度高。当反应中没有甲硫醚和NCS参与时,反应不进行;当甲硫醚与NCS的摩尔量小于2,6-二氟苯胺的0.3倍时,反应时间较长,反应收率和产物纯度较低;当甲硫醚与NCS的摩尔量大于2,6-二氟苯胺的0.7倍时;反应收率和产物纯度也会明显降低。
3.4.2 反应温度对目标产物收率的影响
由表5可知,在其他反应条件不变的情况下,反应温度为40℃,反应时间为4 h,收率和产品含量纯度最高。反应温度越低,反应时间越长,副产物越多;反应温度过高,副产物增加,从而降低了反应收率和产品含量。
4 结 论
实验表明,用2-甲氧基-5-氟尿嘧啶替代5-氟尿嘧啶,经氯化反应、肼基化反应、环合反应、转位反应、偶联反应和氯氧化反应制备关键中间体2-氯磺酰基-8-氟-5-甲氧基[1,2,4]三唑[1,5-c]嘧啶,收率更高,成本更低;磺酰胺缩合反应的最佳工艺条件(摩尔比)为n(2-氯磺酰基-8-氟-5-甲氧基[1,2,4]三唑[1,5-c]嘧啶)∶n(2,6-二氟苯胺)∶n(NCS)∶n(甲硫醚)=1∶1∶0.5∶0.5,缚酸剂用量为2-氯磺酰基-8-氟-5-甲氧基[1,2,4]三唑[1,5-c]嘧啶的3倍量,反应温度为40℃,磺酰胺缩合反应的收率为94.2%。优化后的工艺,以2-甲氧基-5-氟尿嘧啶为起始原料,产品总收率达48.8%,原药含量大于98%。该合成工艺路线更合理,反应条件温和,原料易得,生产成本较低,产品含量高,重现性好,适合工业化生产。
猜你喜欢
含能材料(2022年10期)2022-10-22
云南化工(2022年8期)2022-08-16
纺织检测与标准(2021年3期)2021-12-03
同位素(2021年5期)2021-10-23
农药学学报(2021年4期)2021-08-26
长江大学学报(自科版)(2021年3期)2021-06-01
当代化工(2020年9期)2020-11-30
山西农业科学(2020年11期)2020-11-19
火工品(2019年3期)2019-09-02
染整技术(2019年2期)2019-04-20
