
吹扫捕集-气相色谱质谱法测定水中反式-1,2-二氯乙烯的不确定度评定
2021-03-16 04:01李翠霞
中国新技术新产品 2021年24期
时 妍 李翠霞
(1.吉林市水务集团有限公司,吉林 吉林 132000;2.吉林市职业危害检测检验中心,吉林 吉林 132000)
不确定度是根据所用到的信息来表征赋予被测量量值分散性的非负参数。通过检测分析方法测量不确定度各分量的来源和大小,以评估检测结果的准确性[1]。随着工业的发展,水中污染物的种类,尤其是有机污染物也日益增多。1,2-二氯乙烯有2 个同分异构体,分别为反式-1,2-二氯乙烯和顺式-1,2-二氯乙烯。其中,水中反式-1,2-二氯乙烯主要来自于工业污染,对人体有害[2]。因此,为确保水样检测结果的准确性和可靠性,测量不确定度的评定就显得尤为重要。
目前,检测有机物的方法有很多,其中,吹扫捕集仪简化了样品的前处理过程,使样品分析更加简便,气相色谱质谱联用仪(GC-MS)适合分析易挥发性、分子小、热稳定且能气化的化合物,而且所需试样量少,分离效率高,灵敏度高,具有高选择性的特点,应用范围很广,是唯一可以确定分子式的方法。将吹扫捕集仪和气相色谱质谱联用仪有机结合,可使其检验检测优点最大化。因此,该文采用吹扫捕集-气相色谱质谱法对反式-1,2-二氯乙烯进行检测分析,然后根据《测量不确定度评定与表示》、《化学分析测量不确定度评定》及文献[3]对吹扫捕集-气相色谱质谱法测定水中反式-1,2-二氯乙烯的测量不确定度进行评定,明确了各不确定度分量对检测结果的影响程度,为生活饮用水中挥发性有机物的含量检测方法提供了技术支持。
1 检测方法
1.1 原理
吹扫捕集仪的吹扫管通常用氦气对水样进行吹扫,将反式-1,2-二氯乙烯从水样中吹脱出来,然后被装有吸附剂的捕集阱捕获,通过瞬间加热捕集阱并用氦气反吹,将所吸附的反式-1,2-二氯乙烯解吸入气相色谱质谱联用仪中进行分离测定。根据保留时间和计算机谱库中的质谱图进行定性,再通过定量离子强度和外标法绘制的标准曲线来定量。
1.2 主要仪器和试剂
试验过程中相关仪器和试剂如下:1) 吹扫捕集为Tekmar 9800。2) 气相色谱质谱联用仪为安捷伦7890A-5975C。3) 吹扫捕集进样器容量为5 mL。4) 微量进样器容量为5 μL、25 μL、50 μL、100 μL 以及500 μL。5) 容量瓶容量为100 mL。6) 反式-1,2-二氯乙烯标准溶液购自中国计量科学研究院,溶剂为甲醇,标准值为0.881 mg/mL。7) 纯水为超纯水,空白样品检测无响应。8) 载气为高纯氦气(99.999%以上)。
1.3 仪器参数条件
1.3.1 吹扫捕集条件
吹扫温度为20 ℃,吹扫时间为5 min,解吸温度为170 ℃,解吸时间为5 min,烘烤温度为200 ℃,烘烤时间为5 min,吹扫流量为(40±5)mL/min。
1.3.2 气相色谱质谱联用仪条件
气相色谱仪为DB-5MS 毛细管色谱柱。色谱柱升温程序的起始温度为35 ℃(保持2 min),以10 ℃/min 的速度升温到55 ℃(保持1.5 min),再以30 ℃/min 的速度升温到150 ℃(保持0 min)。柱流量为0.48 mL/min。分流比为20 ∶1;进样口温度为230 ℃。在质谱仪中,离子源(EI)温度为230 ℃,四极杆温度为150 ℃,接口温度为250 ℃,离子化能量为70 eV,扫描范围为35 u~300 u(u 为气质联用仪扫描单位),扫描时间为0.45 s,回扫时间为0.05 s,溶剂延迟时间为2 min。
1.4 水样采集与保存
准备容量为100 mL 的棕色磨口采样瓶,用超纯水清洗采样瓶数次。出厂水的采集需要打开水龙头放水约10 min,达到水温恒定,调节水流的流速为500 mL/min,向采样瓶中以每40 mL 水样加入25 mg 抗坏血酸稳定剂的量来加入相应的稳定剂,将水样充满采样瓶至溢流,旋紧瓶盖;水源水的采集需要用源水冲洗已洗净的采样瓶(数次),将水样充满采样瓶至溢流,旋紧瓶盖。将装有水样的采集瓶放置在4 ℃冰箱低温保存,水样存放区域要保证没有有机物的干扰,在采集后14 d 内对水样进行检测分析。
1.5 操作步骤
根据《生活饮用水标准检验方法》(GB/T 5750.8—2006)有机物综合指标附录A 吹扫捕集/气相色谱质谱法对反式-1,2-二氯乙烯进行测定。按照实验室要求,仪器开机预热至少4 h,以达到完全真空的状态,然后进行GC-MS 性能测试,调谐并分析调谐报告,以满足测定条件,否则重新调谐质谱仪,直到符合要求方可对样品进行分析测定。对高浓度标准储备液进行一级稀释,配制成标准中间液。吸取100 μL 含有(反式-1,2-二氯乙烯)标准物质的储备液加入100 mL 容量瓶中,然后用超纯水定容,混匀后分装到20 mL 顶空瓶中,注意瓶顶上端不留空间和气泡,封盖后可放置4 ℃冰箱中低温保存。在试验测定前,将装有标准中间液的顶空瓶取出,与室温平衡。分别吸取2.5 μL、5.0 μL、20.0 μL、50.0 μL、100.0 μL、250.0 μL 以及500.0 μL 的标准中间液,直接将其注入定容到装有不含有机物超纯水的5 mL 吹扫捕集进样器中,混合均匀后进样测定,即可配制成含有7 个标准浓度点的标准溶液。绘制标准曲线时,标准溶液要现用现配,要使该标准物质的国家标准限值高于标准曲线。每次只配制1 个标准溶液浓度点进行进样分析测定,得到各标准溶液浓度值相对应的响应值,将所得各标准浓度点的数值进行数据处理,用最小二乘法进行拟合,最终得到回归方程和相关系数。
2 数学模型的建立
标准曲线的回归方程如公式(1)所示。
Y=aC+b(1)式中:Y为响应值;C为标准样品的浓度;a为校准曲线的斜率;b为校准曲线的截距。
3 不确定度来源分析
根据整个试验的操作过程可以得出以下4 个方面为测定反式-1,2-二氯乙烯过程中所引入的不确定度的主要来源:1) 配制标准曲线的过程所引入的不确定度urel(C1)。2) 最小二乘法拟合标准曲线的过程所引入的不确定度urel(C2)。3) 重复测量的过程所引入的不确定度urel(C3)。4) 加标回收率的过程中引入的不确定度urel(Rec)。
4 不确定度评定计算依据
该文主要根据《测量不确定度评定与表示》和《化学分析测量不确定度评定》中的方法对整个试验过程中所引入的各不确定度来源进行分析计算。
5 不确定度的评定
5.1 配制标准曲线的过程中引入的不确定度urel(C1)
5.1.1 标准溶液自身所引入的不确定度u(C储)
实验室所用标准溶液是购自有标准物质生产资质的生产厂家,在该文所用反式-1,2-二氯乙烯标准储备液的证书中,已给出相对扩展不确定度,即3%(k=2,k为包括因子),根据不确定度评定计算依据可知,标准溶液自身所引入的相对标准不确定度u(C储)=0.03/2=0.015。
5.1.2 配制标准溶液系列过程所引入的不确定度u(V标)

在下文的相关公式中,ΔV为体积变化允许误差;V为器具体积;ΔT为温度变化允许误差。
5.1.2.1 5 μL 微量进样器所引入的不确定度urel(V5)
5.1.2.1.1 体积引入的不确定度u1(V5)

5.1.2.2 100 mL 容量瓶引入的不确定度urel(V容)
5.1.2.2.1 体积引入的不确定度u2(V容)

根据上述方法可以计算出该实验中各微量进样器、吹扫捕集进样器和容量瓶所引起的标准不确定度和相对标准不确定度,根据绘制标准曲线配制各浓度点的实际过程可知各器具所使用的次数,见表1。
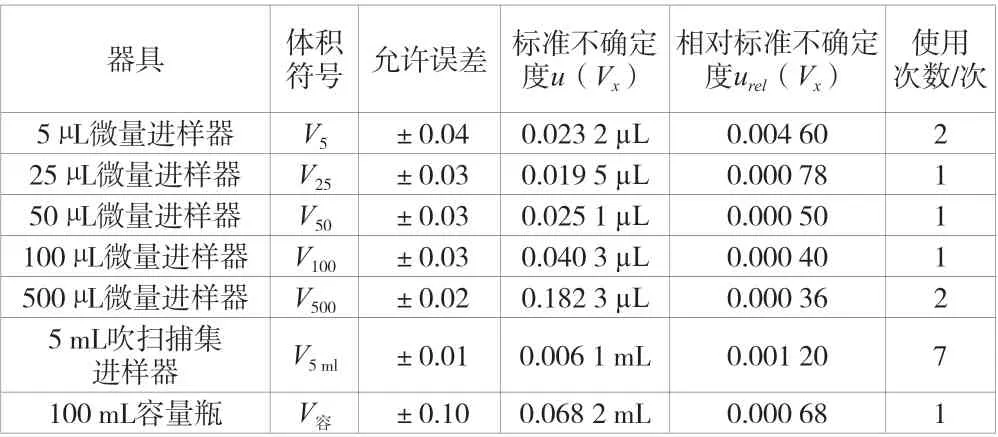
表1 该实验中进样器和容量瓶所引起的标准不确定度和相对标准不确定度
根据上述数据和不确定度评定计算依据可以计算出配制标准溶液系列产生的相对标准不确定度urel(C标)为各项相对标准不确定度的平方与使用次数相乘的和再开平方,计算得urel(C标)=0.007 4。因此,配制标准曲线过程所引入的不确定度urel(C1)为u(C储)和urel(C标)的平方和,再开平方得出urel(C1)=0.016 7。
5.2 最小二乘法拟合标准曲线的过程所引入的不确定度urel(C2)
建立了7 个浓度点的标准曲线,结果见表2。分别取50 μL标准中间液注入装有不含有机物超纯水的5 mL 吹扫捕集进样器中,配置成一定浓度的待测样品并进行7 次独立的重复测定,计算得相对标准偏差,结果见表3。
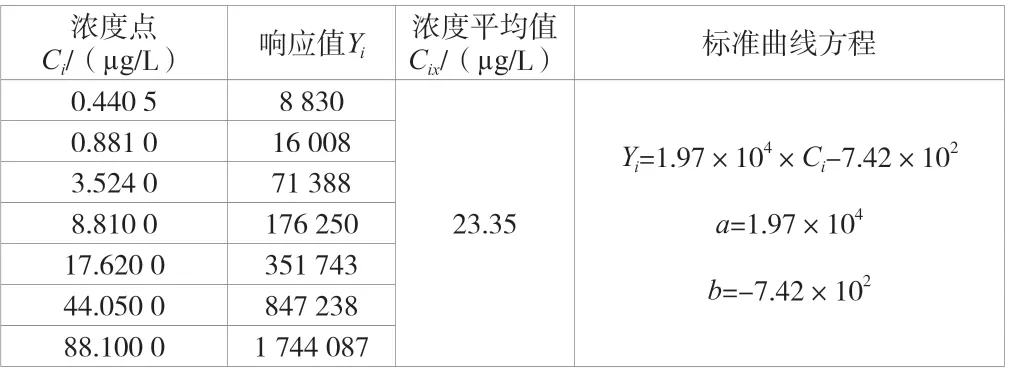
表2 标准曲线各浓度点下的响应值
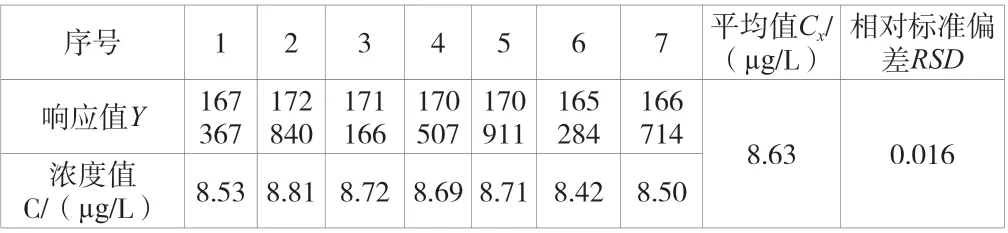
表3 样品测定结果
根据不确定度评定计算依据可查得公式,将标准曲线各值代入公式(1),计算得标准不确定度u(C2)=0.295 8 μg/L。式中:SR为回归曲线剩余标准差(残差的标准差)。
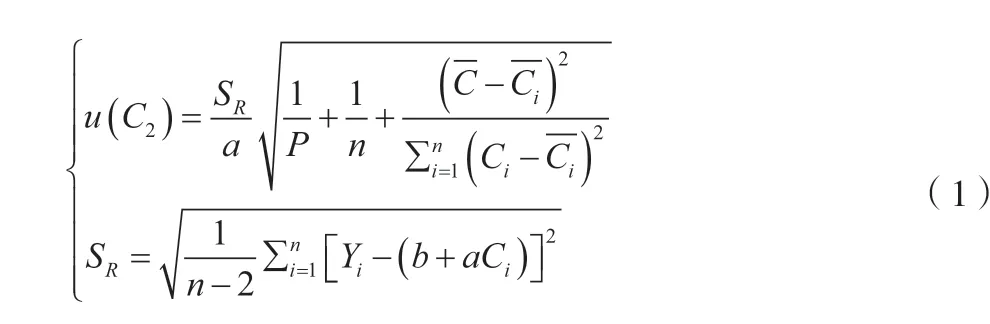
因此,最小二乘法拟合标准曲线的过程所引入的不确定度urel(C2)=0.2958/8.63 ≈0.034 3。
5.3 重复测量的过程所引入的不确定度urel(C3)
对待测样品浓度进行7 次独立重复测定,测定的平均值为Cx=8.63 μg/L,结果为A 类评定,其结果见表3。根据不确定度评定计算依据,计算得标准不确定度u(C3)=0.1423 μg/L。因此,重复测量的过程所引入的不确定度urel(C3)=0.1423/8.63 ≈0.016 5。
5.4 加标回收率的过程所引入的不确定度urel(Rec)
加标回收率引入的不确定度主要来源为吹扫捕集前处理吹扫、转移进入GC-MS 的过程中。向空白水样中分别加入20 μL、50 μL 以及250 μL 的标准中间液,配置成分别含有3.524 μg/L、8.81 μg/L 以及44.05 μg/L 浓度的3 个加标样品,对3 个加标样品进行加标回收率试验并计算得各加标浓度下的加标回收率,测定结果见表4。
由表4 可知,反式-1,2-二氯乙烯的加标回收率为88.7%~95.3%,平均回收率为92.5%。因此,根据不确定度评定计算依据,加标回收率的过程所引入的不确定度urel(Rec)=0.019 1。
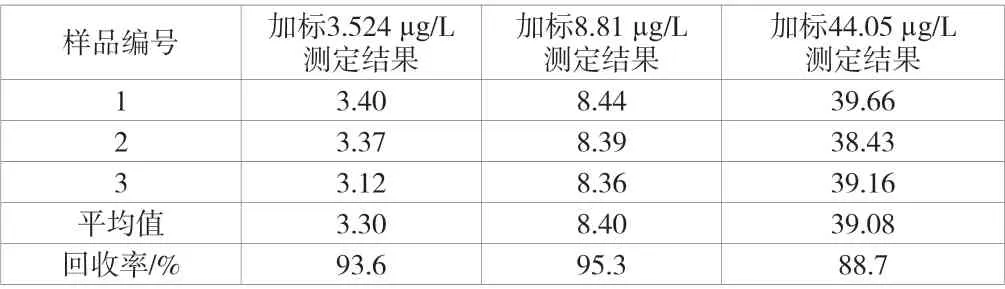
表4 样品加标回收实验测定结果
6 合成标准不确定度U(C)
将所引入的4 个方面的不确定度来源的计算结果汇总,见表5。

表5 相对标准不确定度计算结果汇总
根据检测方法,各不确定度分量之间相互独立,参照不确定度评定计算依据,计算各相对标准不确定度的平方和再开平方得到urel(C)=0.045 7。因此,其合成相对标准不确定度U(C)=urel(C)×Cx=0.0457×8.63 ≈0.394 4 μg/L。
7 扩展不确定度U
一般取包括因子k=2(置信概率为95%),参照不确定度评定计算依据可知,扩展不确定U=U(C)×k,则U=0.3944×2=0.79 μg/L。
8 最后测量结果报告
水中反式-1,2-二氯乙烯含量的测量结果如下。C=(8.63±0.79) μg/L (k=2)
9 结语
该文对吹扫捕集-气相色谱质谱法测定水中反式-1,2-二氯乙烯的测量不确定度评定进行研究。通过表5 可知,对不确定度影响来源由大到小依次为最小二乘法拟合标准曲线、加标回收率、配置标准曲线以及重复测量。其中,最小二乘法拟合标准曲线对不确定度的影响最大,因此,在实验中应注意微量进样器在吸取标准中间液及定容至5 mL吹扫捕集进样器的操作过程,要提高操作的规范性,降低不确定度。同时,该不确定度评定的方法可以为在同方法、同条件水样中其他挥发性有机物的检测提供测量结果不确定度的计算依据,确保实验室分析测试的数据准确性、可靠性。
猜你喜欢
粮食储藏(2023年2期)2023-07-08
中国应急管理科学(2022年2期)2022-05-23
中国生殖健康(2020年5期)2021-01-18
中国盐业(2018年16期)2018-12-23
中国生殖健康(2018年5期)2018-11-06
食品与机械(2018年5期)2018-07-16
中国民族医药杂志(2016年5期)2016-05-09
化工生产与技术(2014年4期)2014-02-27
化学分析计量(2011年1期)2011-04-11
