
基于近红外光谱的大米蛋白质含量快速检测
2021-06-04 09:19刘金明张东杰34张爱武3
食品与机械 2021年5期
殷 坤 刘金明 张东杰34 张爱武3
(1.黑龙江八一农垦大学食品学院,黑龙江 大庆 163319;2.黑龙江八一农垦大学信息与电气工程学院,黑龙江 大庆 163319;3.黑龙江八一农垦大学国家杂粮工程技术研究中心,黑龙江 大庆 163319;4.黑龙江省农产品加工与质量安全重点实验室,黑龙江 大庆 163319)
大米蛋白质是公认的优质植物蛋白质之一[1],其氨基酸组成均衡合理,不会产生过敏反应[2]。研究[3]表明,大米中蛋白质含量与大米的食味品质呈负相关,蛋白质含量增加,会导致米饭的硬度增大、弹性降低、色泽变差,大米的食味品质变差。大米蛋白质含量的检测是评价大米质量的重要手段之一。目前,蛋白质含量的测定方法主要依据GB 5009.5—2016《食品安全国家标准 食品中蛋白质的测定》中的凯氏定氮法、分光光度法和燃烧法。该国标检测方法具有准确性好、灵敏度高的特点,但操作繁琐、检测时间长且破坏样品。
近红外光谱(near infrared spectroscopy,NIRS)分析技术因其简便、快速、低成本、无损以及多组分同步检测的优点被广泛应用于大米蛋白质、脂肪等成分含量的快速检测。苗雪雪等[4]使用偏最小二乘(partial least squares,PLS)、组合区间PLS和移动窗口PLS 3种方法对327份大米样本的NIRS和水分含量进行建模,结果表明移动窗口PLS法建立的模型的预测集相关系数达到了0.961 7,可以用于大米水分含量的快速检测。刘文丽等[3]使用OPUS定量分析软件分别对大米蛋白质和蛋白质(干基)进行PLS建模,得到的最佳近红外模型决定系数R2分别为0.956 5和0.974 1,剩余预测偏差(residual predictive deviation,RPD)分别为4.79和6.21,说明两种模型都有较好的精确性,可以用于预测大米中蛋白质含量。俞法明等[5]对同一批样品的稻谷、糙米、精米、精米粉分别建立了蛋白质含量NIRS检测模型,发现精米和精米粉回归模型的校正决定系数大于0.90,可用于进行大米蛋白质含量的快速检测。
上述大米组分NIRS快速检测模型主要采用PLS线性建模方法建立相应的校正模型,但PLS难以体现光谱数据与待测目标组分之间的非线性关系。因此,相关学者开始研究使用支持向量机(support vector machine,SVM)和神经网络等非线性多元统计方法建立NIRS定量回归模型[6]。其中,SVM是基于统计学习理论和结构风险最小化原则的机器学习方法[7],与传统的神经网络相比结构简单、泛化能力强,对求解小样本、非线性、高维数问题有更好的适应性。但是在应用SVM进行光谱数据非线性建模时,因NIRS原始数据维数过高,若直接以全部的波长数据作为SVM的输入变量,将导致模型过于复杂、运行时间长,而且冗余光谱数据还会影响模型的建模精度[8]。李子文等[9]分别使用PLS和最小二乘SVM建立了372个核桃露样品脂肪含量NIRS定量检测模型,结果表明两种模型均有较高的精度,且最小二乘SVM建立的模型性能更高。孙丽萍等[10]使用SVM建立了东北黑木耳多糖含量的NIRS分类模型,模型精确率为85.7%,召回率为87.3%,说明SVM可以用来建立黑木耳多糖含量分类模型。
相关学者将主成分分析(principal component analysis,PCA)与SVM相结合构建PCA-SVM用于建立NIRS快速检测模型。张晓冬等[11]使用PCA-SVM建立了4种含铁矿物药的NIRS快速鉴别模型,结果表明所建的模型预测能力强,可以用于4种含铁矿物药的快速鉴别。魏从师等[12]利用NIRS法结合PCA和SVM建立了6味中药的识别模型,所建的模型对训练集和验证集样品预测的正确率均达到100%。上述研究均使用网格搜索(grid search,GS)对SVM建模参数进行优化,但GS法搜索步长过大时结果不准确,搜索步长小则运行时间较长[13]。而且前人的研究大多基于全谱进行建模,在特征波长优选与PCA-SVM结合方面少有涉猎。研究拟基于遗传模拟退火算法(genetic simulated annealing algorithm,GSA)对PCA-SVM的参数进行优化,探讨将PCA-SVM与反向区间偏最小二乘(backward interval partial least squares,BiPLS)相结合建立大米蛋白质含量NIRS快速检测模型的可行性,以BiPLS优选后的谱区作为PCA-SVM的输入参数,建立大米蛋白质含量快速检测模型。
1 材料与方法
1.1 材料与仪器
1.1.1 材料与试剂
大米:采集自黑龙江省查哈阳、五常、方正、响水和建三江5个地区150个样品,垄谷3次、碾米2次后粉碎,装入密封袋于阴凉干燥处保存;
硫酸、硫酸钾、硼酸、乙醇:分析纯,辽宁泉瑞试剂有限公司;
硫酸铜:分析纯,哈尔滨市新达化工厂;
氢氧化钠:分析纯,天津市博华通化产品销售中心;
甲基红、溴甲酚绿:分析纯,天津市恒兴试剂制造有限公司。
1.1.2 主要仪器设备
垄谷机:THU35C型,佐竹机械(苏州)有限公司;
碾米机:TM05C型,佐竹机械(苏州)有限公司;
粉碎机:CT193型,福斯分析仪器(苏州)有限公司;
电子分析天平:AR323CN型,奥豪斯仪器(上海)有限公司;
全自动凯氏定氮仪:K9860型,济南海能仪器股份有限公司;
石墨消解仪:SH220N型,济南海能仪器股份有限公司;
傅里叶变换近红外光谱仪:TANGO型,德国BRUKER公司。
1.2 方法
1.2.1 蛋白质含量测定 按GB 5009.5—2016的凯氏定氮法执行。
1.2.2 光谱数据采集 粉碎后的大米样品填满样品杯,振荡均匀后使用近红外光谱仪进行积分球漫反射光谱扫描,光谱范围3 946~11 542 cm-1(866~2 534 nm),分辨率为8.0 cm-1,每个样品扫描32次,背景每小时扫描一次。保持室内温湿度基本不变,每个样品装样3次,取3次扫描平均值作为样品原始光谱。
1.2.3 BiPLS BiPLS是在间隔偏最小二乘法(interval partial least square,iPLS)[14]的基础上发展而来的一种特征谱区优选算法。BiPLS在iPLS的基础上依次去除交叉验证均方根误差(root mean squared error of cross-validation,RMSECV)值最大的区间,当RMSECV最小时所对应的子区间组合为优化后的特征谱区域[15]。
1.2.4 PCA-SVM PCA作为一种多元统计方法,能够消除各指标间的相互关联影响,在数据降维方面应用广泛[16]。SVM非线性回归的核心思想是通过选取的核函数将非线性问题映射为高维特征空间的线性问题,并在这个空间中进行线性回归[17]。
PCA-SVM融合了PCA光谱数据降维方法和SVM非线性回归思想,既解决了PLS在处理光谱数据多重共线性时效果不佳的问题,又弥补了以全谱波长变量直接建模效率低的不足。在采用径向基(radial basis function,RBF)核函数进行PCA-SVM建模过程中,主成分(principal components,PCs)个数、惩罚参数C、RBF核函数参数γ和不敏感损失函数参数ε对建模性能具有重要影响。采用MCCV结合PLS回归模型进行最佳PCs个数的选取。通过比较PLS回归模型的预测残差平方和(prediction residual error sum of squares,PRESS),选取PRESS值最小的PCs个数作为最佳PCs个数[18]。为了有效提高PCA-SVM的学习和泛化能力,采用GSA算法[19]对回归模型的参数C、γ和ε进行优化。GSA以PCA-SVM回归模型的k折RMSECV为目标函数,结合温度参数进行适应度优化设计如下:
(1)
式中:
fit(x)——适应度函数;
f(x)——当前染色体目标函数值;
fmin——当前代种群中的最小目标函数值;
t——当前代温度值。

(2)
(3)
(4)
式中:
R2——决定系数;
RMSE——均方根误差;
RPD——剩余预测偏差;
yi——第i个样本的测量值;


n——样本个数。
研究所有算法,包括光谱预处理、样本集划分、特征谱区优选、主成分分析、模型参数优化和回归模型构建等全部在Matlab R2016b软件平台中实现,其中BiPLS和SVM分别基于iToolBox[21]和LibSVM[22]工具箱实现。
2 结果与分析
2.1 采集数据分析
大米样品光谱数据如图1所示。由图1(a)可知,原始光谱数据存在基线漂移现象。由图1(b)可知,通过SG平滑和MSC多元散射校正相结合对原始光谱进行预处理,修正了因扫描时间、周边环境、设备状态不同而导致的基线漂移、噪声干扰和光谱散射等问题,有效提高了光谱数据的分辨率和信噪比。其中,SG主要用于消除随机噪声的干扰,MSC主要用于修正基线漂移和光谱散射。对NIRS数据进行预处理后,使用KS法按4∶1的比例进行样本集划分,得到校正集样本120个、验证集样本30个。样品中蛋白质含量测量结果如表1所示。
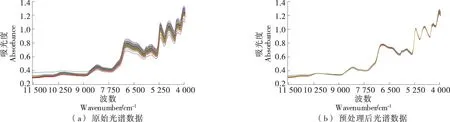
图1 样品光谱数据Figure 1 Spectral data of samples
对预处理后的NIRS数据进行PCA计算,第一、第二和第三PCs的贡献率分别为82.72%,7.31%,6.07%,前3个PCs的累积贡献率达96.10%。校正集和验证集的三维PCs空间分布情况如图2所示。
由表1和图2可知,校正集基本涵盖了验证集,且校正集和验证集样本在PCs空间上分布均匀,可以用该样本划分方法进行NIRS建模。

图2 样本在主成分空间中的分布Figure 2 Distribution of the samples in principal components space

表1 样品中蛋白质含量Table 1 Protein content in samples
2.2 特征谱区优选
在使用BiPLS算法进行NIRS特征谱区优选时,区间划分个数的选择很重要。为寻找最佳区间划分个数,首先将预处理后的样品光谱数据划分为k个子区间(k=10,20,30,40,50,60,70,80,90),并采用BiPLS计算每个k值下的最佳特征谱区,完成划分区间个数的初选(如表2 所示)。由表2可知,k=70时,蛋白质BiPLS回归模型的RMSECV最小;k=80时,其RMSECV比k=70时略大,因此可以推测蛋白质最佳谱区划分个数为70~80。
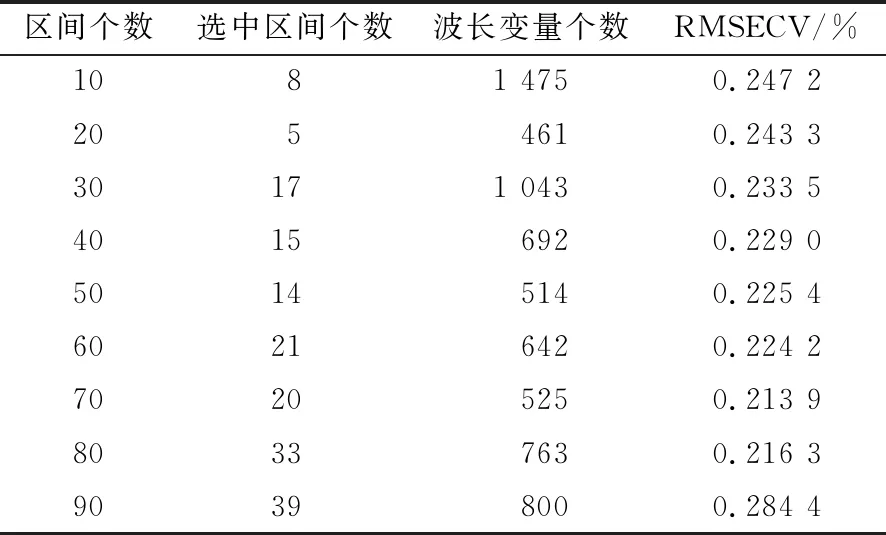
表2 BiPLS优化蛋白质光谱特征区间初选结果Table 2 Primary results of BiPLS optimization of protein spectral feature interval
为获得蛋白质最佳谱区划分个数,采用BiPLS计算区间个数从71到79对应的最佳特征谱区(如表3所示)。由表3可知,区间划分个数为79时,BiPLS优选出42个蛋白质特征谱区对应的RMSECV最小(0.211 0),对应区间列表为[1 3 4 7 8 9 10 12 15 17 19 20 22 23 25 26 27 30 31 32 33 34 36 37 39 40 44 46 47 48 50 57 58 63 64 68 69 70 72 74 76 77],选中的特征波长变量共983个。

表3 BiPLS最佳特征谱区筛选结果Table 3 Results of BiPLS optimal feature spectrum screening
绘制BiPLS优选的蛋白质特征谱区与光谱数据对比图(如图3所示)。图3中,特征谱区波数9 075~9 450 cm-1区域对应着蛋白质—RNH2基团的三级倍频;波数6 900~7 300 cm-1区域对应着蛋白质—CONH2基团的二级倍频;波数5 640~5 880 cm-1对应着蛋白质—CH和—CH2基团的一级倍频,波数4 220~4 370 cm-1区域对应着蛋白质—CH、—CH2和—CH3基团的组合频。
2.3 回归模型建立及评价
最佳PCs个数的选择是建立PCA-SVM模型的第一步,合理的PCs个数既可以提高模型的预测精度,又可以提高模型的稳定性。采用MCCV结合PLS回归模型进行最佳PCs个数的选取。设PCs个数的搜索范围为1~20,步长为1,分别建立不同PCs个数下的PLS回归模型,并计算每个PLS回归模型的PRESS,选取PRESS值最小时对应的PCs个数作为最佳PCs个数。以BiPLS优选的983个蛋白质特征波长变量为输入,采用MCCV结合PLS回归模型分析PRESS随PCs个数变化情况如图4 所示。由图4可知,随着PCs个数的增加,PRESS呈先迅速减少,后缓慢增加的趋势。图4中箭头所示红色圆点的PRESS值最小(2.182 6),PCs的个数为11,说明这11个PCs的解释能力最强。选取此点的PCs个数作为大米蛋白质BiPLS特征波长优选后的最佳PCs个数建立PCA-SVM模型。
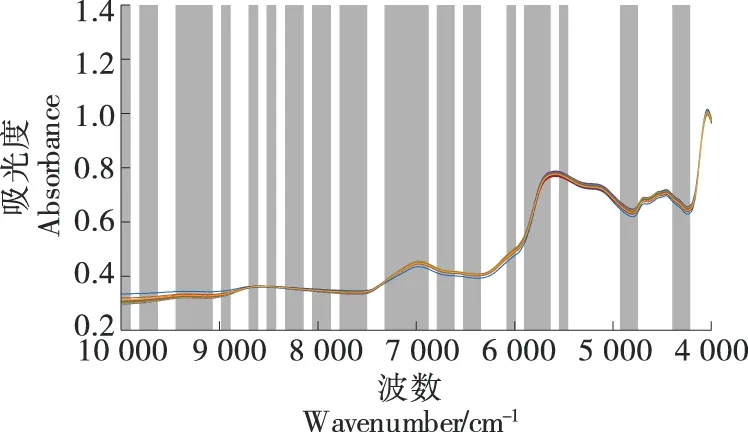
灰色阴影区域为蛋白质特征谱区图3 蛋白质光谱特征谱区Figure 3 Spectral characteristic intervals of protein

图4 PRESS与PCs关系图Figure 4 Relationship between PRESS and the number of PCs
遗传算法(genetic algorithm,GA)因其具有强大的搜索能力,在SVM参数优化方面得到了广泛应用[23]。但GA存在早熟问题,且进化后期搜索效率较低。因此,采用GSA算法对PCA-SVM回归模型的参数C、γ和ε进行优化。参数C、γ和ε的寻优范围分别是[0,100]、[0,100]和[0.001,1.000],染色体码长为60(每个参数对应20位基因),种群规模为50,遗传代数为200,初始温度参数K为200,退温系数α为0.95,交叉概率为0.7,变异概率为0.7/L(L为染色体码长),采用10折RMSECV为目标函数。以BiPLS优选的983个大米蛋白质特征波长变量按最佳PCs个数为11,使用校正集数据进行PCA-SVM回归模型参数优化。参数优化的过程如图5所示。由图5 可知,在高温时(进化前期)GSA求得的平均目标函数值与最佳目标函数值差异很大,而低温时(进化后期)平均目标函数值更接近于最佳目标函数值。原因在于GSA算法融合了GA和模拟退火算法的优点,结合温度参数设计适应度函数和Metropolis选择复制策略,既提高了算法高温时(进化前期)种群的多样性,又扩大了低温时(进化后期)优良染色体的适应度函数值,能够有效克服GA早熟收敛和进化后期搜索效率低的不足。
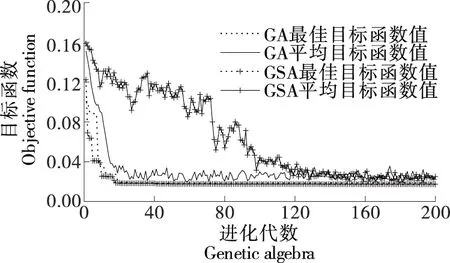
图5 基于特征波长的蛋白质PCA-SVM的参数优化过程Figure 5 Optimization process of parameters for PCA-SVM based on characteristic wavelengths for protein
为了分析BiPLS与PCA-SVM相结合建立大米蛋白质含量NIRS快速检测模型的性能,分别建立全谱PLS和BiPLS优选谱区的PLS、SVM和PCA-SVM回归模型,依次记为Full-PLS、BiPLS、BiPLS-SVM和BiPLS-PCA-SVM。不同回归模型的评价指标如表4所示。


表4 不同回归模型的评价指标Table 4 Evaluation indicators of different regressive models
由图6可知,蛋白质的测量值与预测值点基本呈对角线分布,样本点位于对角线两侧,说明蛋白质回归模型的预测效果较好。经t检验计算,得到样本蛋白质含量实测值与预测值的t值为1.058 2,小于相应的临界值t(0.05,149)=1.976,Matlab ttest函数返回检验结果均为0,显著性水平概率为0.928 2,说明蛋白质测量值与预测值无显著性差异,一致性较好。图6中存在个别偏离对角线较严重的样本点,原因在于部分样品消化过程中因淀粉含量过高,导致在消化温度300 ℃左右时产生大量黑色气泡粘壁,消化温度提高到420 ℃后部分附着在消化管壁的气泡无法被消化,从而形成未消化完全的黑色颗粒物而产生的误差。因此在建立NIRS快速检测模型时,一定要保证前期测量的基础目标属性化学含量值的准确。BiPLS-PCA-SVM融合了BiPLS特征谱区优选、PCA光谱数据降维、MCCV的最佳主成分选取、SVM非线性建模、GSA参数优化,既减少了SVM模型的输入数据量,又提高了SVM模型的回归精度,还有效解决了线性方法建模的多重共线性问题。在建立基于BiPLS-PCA-SVM的大米蛋白质含量NIRS快速检测模型过程中,并未对图6中偏离对角线的异常样本进行剔除,但建立的模型仍能满足大米蛋白质含量的快速检测需求。说明使用BiPLS-PCA-SVM建立NIRS检测模型时,在保证足够多样本的前提下,少量样本化学含量测定存在一定误差对最终模型的影响不大,充分体现了文中提出的BiPLS-PCA-SVM建模方法具有较高的鲁棒性。

图6 测量值和预测值分布图Figure 6 Distribution of measured and predicted values
3 结论
试验探讨了近红外光谱结合化学计量学方法进行大米蛋白质含量快速检测的可行性。为了提高近红外光谱回归模型的检测精度和效率,将反向区间偏最小二乘(BiPLS)、主成分分析(PCA)和支持向量机(SVM)相结合构建BiPLS-PCA-SVM模型,所建立回归模型的验证决定系数为0.928 9,预测均方根误差为0.196 7%,剩余预测偏差为4.024 6。结果表明,BiPLS-PCA-SVM模型通过融合了特征谱区优选、光谱数据降维和非线性建模策略,其建模性能优于全谱偏最小二乘模型和反向区间偏最小二乘优选谱区建立的偏最小二乘和支持向量机回归模型,能够满足大米蛋白质含量的实际检测需求。BiPLS-PCA-SVM与近红外光谱相结合为建立简单、高效、低成本的大米蛋白质含量快速检测方法提供了新途径。由于时间有限,试验仅将BiPLS-PCA-SVM应用于大米蛋白质含量的测定,后期将进一步研究其在大米直链淀粉、脂肪等其他营养成分含量测定方面的应用。
猜你喜欢
北京航空航天大学学报(2022年8期)2022-08-31
肝博士(2022年3期)2022-06-30
小学生学习指导(低年级)(2021年9期)2021-10-14
海外星云(2021年9期)2021-10-14
空间科学学报(2021年1期)2021-05-22
中学生数理化·七年级数学人教版(2019年10期)2019-11-25
小学生学习指导(低年级)(2019年9期)2019-09-25
小学生学习指导(低年级)(2018年9期)2018-09-26
航天返回与遥感(2014年4期)2014-07-31
食品工业科技(2014年23期)2014-03-11
