
Ti3C2电极材料的密度泛函理论计算
2022-01-21 13:20王思奇杨鑫林佘维汉杨光敏
吉林大学学报(理学版) 2022年1期
司 雪, 李 卓, 王思奇, 杨鑫林, 佘维汉, 徐 强, 杨光敏
(1. 长春师范大学 物理学院, 长春 130032; 2. 长春工程学院 勘查与测绘学院, 长春 130012)
近年来便携式电子设备的功耗逐渐增大, 环境污染与全球能源危机日益加剧, 新能源交通工具高速发展, 因此探索清洁、 可持续、 可再生能源已成为该领域目前的研究热点[1-2]. 超级电容器作为新型绿色环保储能装置具有高功率密度、 高可靠性、 体积小、 绿色环保等优点[3-9], 但其能量密度低, 限制了应用. 为提高超级电容器的性能, 寻找优异的电极材料已引起人们广泛关注.
一种新型过渡金属碳化物、 氮化物或碳氮化物二维纳米材料MXenes已应用于超级电容器中, 其中Ti3C2通过蚀刻MAX相中Al原子得到. MAX通式为Mn+1AXn, M和A分别表示过渡金属元素及ⅢA和ⅣA主族元素, X为碳或氮元素,n=1~3. 实验上通常用氢氟酸(HF)蚀刻Ti3AlC2, 其方程为Ti3AlC2+3HF=AlF3+3/2H2+Ti3C2. 在HF和H2O同时存在的条件下, 由于Ti3C2层表面暴露的Ti原子最终端接F—等基团或认为Ti3AlC2的Al层被F—等基团取代, 因此纯MXenes材料在现实中无法存在, 多以表面携带H—,—O—,F—等端基官能团的形式存在. 本文基于密度泛函理论的第一性原理, 研究H—,—O—,F—三种基团对Ti3C2的电子结构和量子电容的调制效果.
1 计算方法和结构模型
1.1 计算方法
计算均采用密度泛函理论的缀加投影波方法, 使用Vienna Ab-initio Simulation Package (VASP)软件包. 电子交换关联能使用Perdew-Burke-Ernzerhof (PBE)泛函. 截断能为450 eV, 层间距设为1.8 nm,K点设置为0.189. 将自旋计算考虑在内.
1.2 结构模型
Ti3C2单层按Ti—C—Ti—C—Ti的顺序堆叠而成, 可描述为3个Ti原子层与2个C原子层形成边缘共享的八面体, 如图1(A)所示. 氟化的Ti3C2通过F—基团使表面欠配位的Ti原子达到饱和而构造. 本文考虑F—等基团3种可能的吸附位置: Ⅰ位于3个相邻C原子的中心处, 直接指向Ti原子位置的正上方; Ⅱ位于两侧C原子层的正上方; Ⅲ位于两侧Ti原子层的正上方, 如图1(B)所示. 本文以最稳定的位置吸附基团, 调制Ti3C2的电子结构, 改善其量子电容.
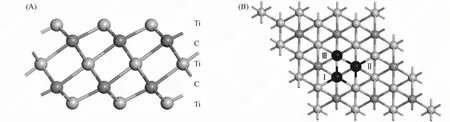
图1 Ti3C2的侧视图(A)及Ti3C2的俯视图和3个可能的吸附位置(B)Fig.1 Side view of Ti3C2(A) and top view of Ti3C2 and three possible adsorption positions (B)
2 结果与讨论
2.1 吸附能
为分析吸附基团对结构稳定性的影响, 分别计算Ⅰ,Ⅱ,Ⅲ三种不同吸附位置环境调制后结构的吸附能, 结果列于表1. 计算吸附能公式为
Ead=Etotal-Epristine-nμF(H,O),
其中Ead,Etotal,Epristine分别为体系的吸附能、 调制后结构的总能量和本征Ti3C2的能量,n为基团数量,μ表示不同基团的能量.
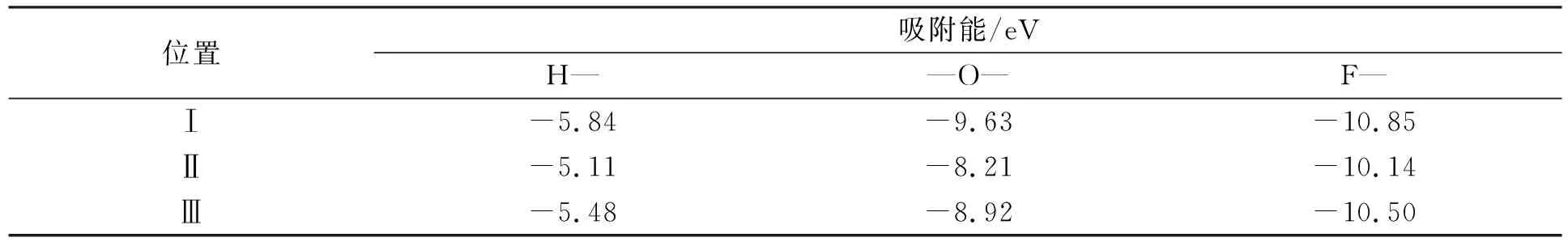
表1 H—,—O—,F—基团在3种吸附位置下的吸附能
由表1可见, H—,—O—,F—三种基团吸附能的最小值分别为-5.84,-9.63,-10.85 eV, 且吸附能均为负值. 表明H—,—O—,F—三种基团在位置Ⅰ吸附时具有最稳定的热力学结构. 因此计算均在最稳定构型的吸附位置Ⅰ进行.
2.2 电子结构调制
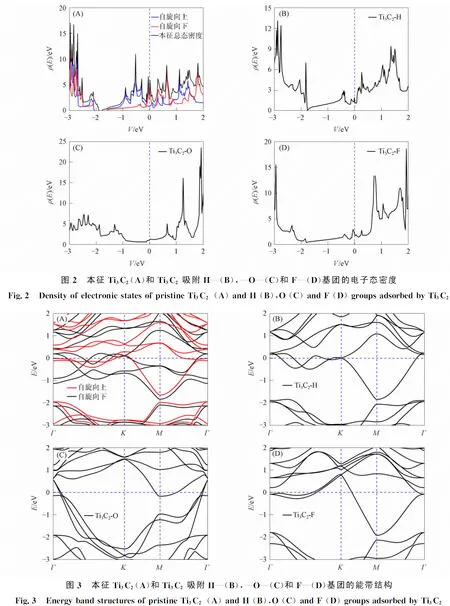
通过对优化结构的态密度和能带进行计算, 分析其电子结构变化, 结果分别如图2和图3所示. 由图2可见: Ti3C2存在自旋, Ti3C2的Ti原子轨道主要分布在-1~2 eV间; Ti3C2在Fermi能级位置表面存在未极化的Ti 3d轨道电子, 因此Ti3C2具有还原性, 从而对H—,—O—,F—基团有较高的吸附活性; 4种结构的态密度在Fermi能级左侧均呈下降趋势, 出现赝能隙, 表明占据上方的电子呈非定域化, 证明这些结构具有金属性. 由图3可见, 4种结构在Fermi能级附近均未出现明显带隙, 且导带和价带在Fermi能级处有明显的重叠部分, 表明它们是具有金属性的材料.
2.3 量子电容与表面电荷密度
Ti3C2在剥离过程中会端接H—,—O—,F—等基团, 通过MATLAB拟合Ti3C2吸附H—,—O—,F—基团的量子电容和表面电荷密度与电势的关系, 结果如图4所示. 由图4(A)可见, Fermi能级附近吸附H—,—O—,F—基团的Ti3C2的量子电容最大值分别为380.01 μF/cm2(-0.288 V), 119.44 μF/cm2(-0.072 V)和228.41 μF/cm2(0.048 V). 因此, Ti3C2吸附H—基团的量子电容性能最好. 量子电容的提高可改善总界面电容, 从而提高超级电容器的性能. 由图4(B)可见, 在正偏压下, Ti3C2吸附H—和F—基团电荷积累效果呈相同的趋势, 在负偏压下, Ti3C2吸附H—基团电荷积累效果最好.
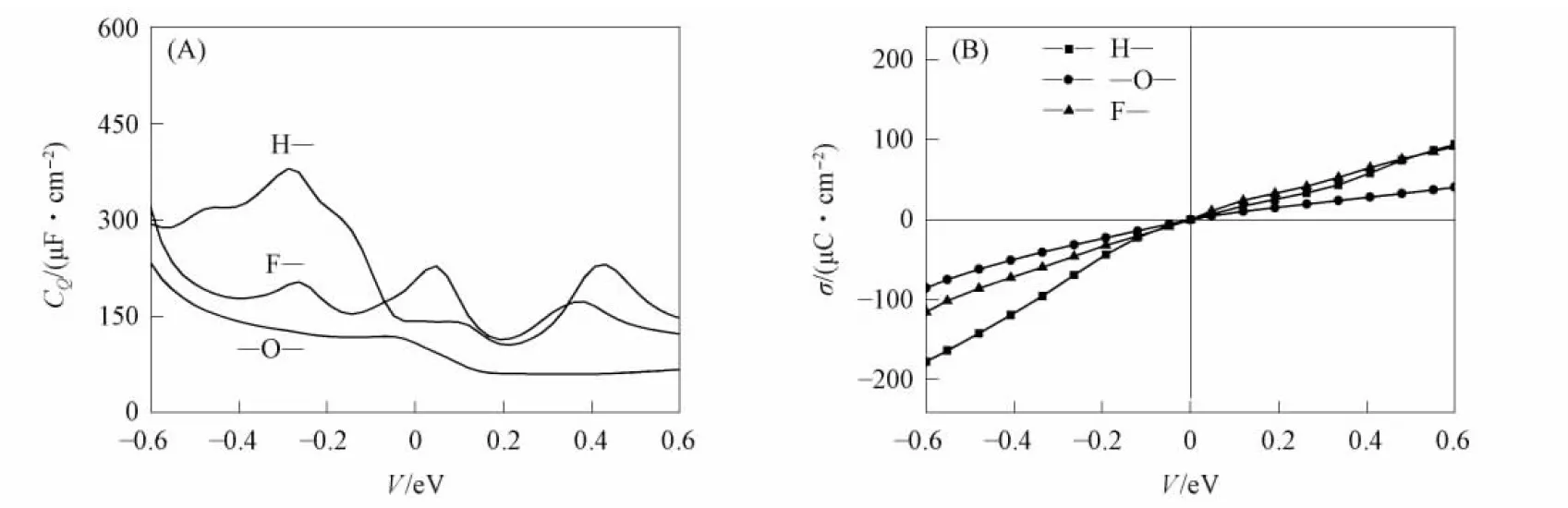
图4 Ti3C2吸附H—,—O—,F—基团的量子电容(A)和表面电荷密度(B)与电势的关系Fig.4 Relationship between quantum capacitance (A), surface charge density (B) of H,O,F groups adsorbed by Ti3C2 and potential
综上, 本文基于第一性原理, 模拟计算了H—,—O—,F—基团修饰Ti3C2的吸附能、 电子性质和量子电容. 结果表明: 在3个相邻C原子中心处的正上方是最佳吸附位; H—,—O—,F—基团最稳定位置的吸附能分别为-5.84,-9.63,-10.85 eV; 4种材料均呈明显的金属性; Ti3C2吸附H—基团的量子电容性能最好, 且在负偏压下, Ti3C2吸附H—基团的电荷积累效果最好.
猜你喜欢
当代党员(2022年9期)2022-05-20
小作家报·教研博览(2022年11期)2022-04-02
当代陕西(2022年1期)2022-03-09
华人时刊(2021年23期)2021-03-08
科学导报(2020年68期)2020-11-09
新课程·下旬(2019年7期)2019-09-17
学生导报·东方少年(2019年11期)2019-06-11
发明与创新·中学生(2017年11期)2017-12-07
中学生数理化·高三版(2017年1期)2017-04-20
新高考·高一物理(2016年7期)2017-01-23
