
超声诊断孕早期Cantrell五联征并UGT1A1基因复合杂合突变1例病例报告
2022-05-24 02:42郑玉兰唐中锋徐生芳
中国循证儿科杂志 2022年2期
郑玉兰 杨 磊 唐中锋 徐生芳
1 病例资料
女,31岁,因“停经12周”于甘肃省妇幼保健院(我院)产前诊断中心行孕早期常规胎儿颈项透明层(NT)超声筛查,发现胎儿畸形,收入院。孕12周,孕前月经规律,自然受孕,孕期未服用任何药物。G3P0,G1因主观原因行人工流产,G2因“宫角妊娠”行人工流产。停经约8周时发现阴道少量出血,于当地医院超声检查,提示未见异常,予口服黄体酮治疗。既往患有胆囊结石2年余,否认乙肝、结核等传染病史,否认高血压、糖尿病、肾病等慢性病史,否认家族性遗传病史。
入院查体:血压126/83 mmHg,血常规正常,腹部常规超声提示胆囊结石。产前超声提示:胎儿顶臀径60 mm,心率160次/min,胎盘位于前壁,羊水深度23 mm;胎儿胸腹壁连续性回声中断,宽10 mm,脏器从缺损处外翻腹腔外,自由漂浮在羊水中,表面未见膜状物包裹,范围21 mm×20 mm×12 mm;心脏位置下移外翻,结构显示不清(图1A);脊柱排列紊乱弯曲(图1B);颅骨光环完整,脑中线可见,脉络丛不对称;右侧上肢未显示,左侧肢体及双下肢可见;仅见1条脐动脉。超声诊断:宫内单胎妊娠,胎儿存活,孕12+3周;胎儿胸腹裂、脊柱异常、颅脑异常,考虑Cantrell五联征;胎儿右上肢缺如;单脐动脉。
孕妇及其丈夫经产前咨询,认为无法承担围手术期风险等因素,决定引产。引产后大体结果显示:胸腹前见一巨大包块,内可见部分心脏略外翻,可见肝脏、胃泡及部分肠管(图1C),未见膜状物包裹;脊柱弯曲,右上肢缺如(图1D),口唇、其余肢体未见明显畸形。
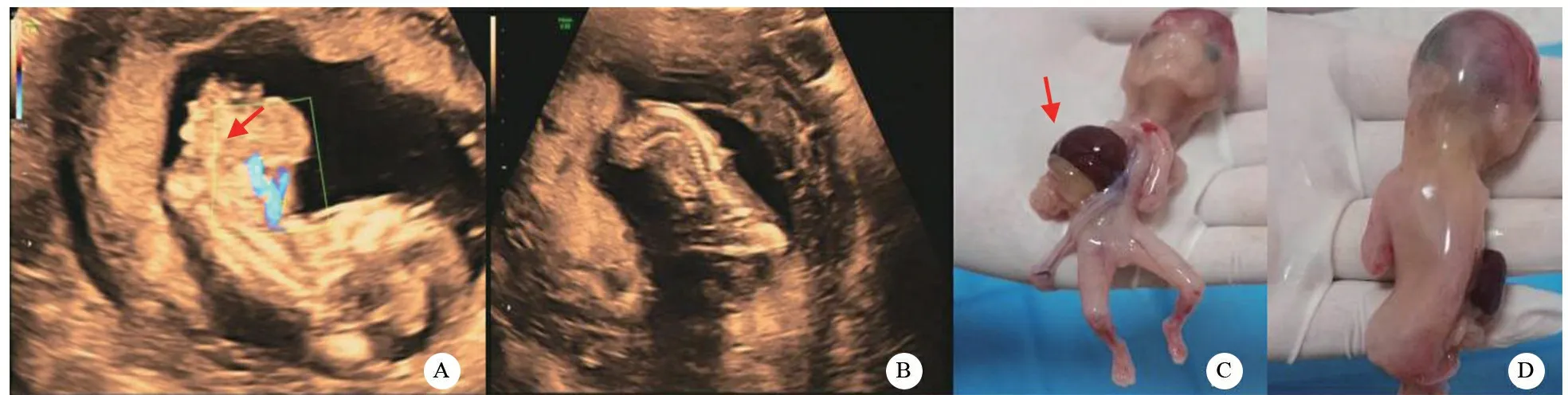
图1 胎儿产前超声和引产后大体观
经孕妇及其丈夫书面知情同意后依次行染色体核型分析、拷贝数异常及家系基因检测,采集胎儿脐带(近胎儿段)2~3 cm,父母外周静脉抗凝血各3 mL,对胎儿及其父母基因行Trio全外显子组测序(WES),捕获芯片为IDT-The-xGen-Exome-Research-Panel-v1.0,用Illumina-NovaSeq-6000系列测序仪行高通量测序,BWA软件进行生物学信息对比分析,对可疑致病变异进行胎儿父母的Sanger测序验证。检测到胎儿携带致病性证据充分的UGT1A1基因(NM_00463)的2个突变位点[C.1456T>G(p.Y486D)、C.1091C>T(p.P364L)],分别来自父亲和母亲,根据人类基因组变异软件PROVEAN,判定为均可能致病。
2 讨论
本文病例行NT常规超声检查时考虑Cantrell五联征。Cantrell五联征是由脐膨出、心脏异位或心内畸形、下部胸骨缺损、膈肌前部及心包缺损5种畸形组成的一种极为罕见的先天性畸形,最早于1958年由Cantrell等提出,发生率为1/200 000~1/65 000,生存率极低[1],很少有患儿能在生命的早期存活下来。此病预后不良,患儿出生后多首选手术治疗,预后取决于胸腹壁缺损的大小及心内结构异常的程度,经手术治疗后也可能发生心力衰竭、败血症、缺氧等并发症而死亡,存活率低至37%[2]。其发病机制尚不明确,多认为是胚胎早期中胚层移行发育缺陷或突变所致。
产前超声对孕早期胸骨异常、心包及膈肌缺损等结构畸形难以直观显示,妊娠10周发现胸外心和脐膨出典型超声特征即可明确诊断Cantrell五联征[5],此外,合并颜面(唇腭裂)、颅脑(全前脑)、脊柱(脊柱侧弯)、肢体(手足畸形、肢体缺)异常,或发育不良,或脐带异常(单脐动脉和/或短脐带等[6])可辅助诊断。产前超声可尽早筛查胎儿Cantrell五联征并行产前咨询。
文献报道[3,4],Cantrell五联征男性较女性多见,与染色体异常有关,如18三体、21三体、特纳综合征或X染色体上的基因突变。有报道认为与15q21.3ALDH1A2基因(在心脏与膈肌发育中起关键作用)、Xp11.23PORCN基因(可能与胸骨缺损有关)和11q14.1TENM4基因异常有关[2-4],目前尚未发现与上述基因相同的变异。
尿苷二磷酸葡萄糖醛酸转移酶lAl(UGT1A1)是一种位于染色体2q37.1上的UGT1A1基因编码的533个氨基酸组成的蛋白质,是人类胆红素葡萄糖醛酸化的唯一酶,该基因包括1个启动子TATA盒序列和5个外显子,突变可引起UGT1A1活性降低或完全丧失[7],导致一系列非结合胆红素升高的疾病,均为常染色体隐性遗传病。本例胎儿组织染色体核型分析正常,染色体拷贝数变异检测结果显示正常,WES测序发现该胎儿携带致病性证据充分的2q37.1UGT1A1基因的点突变,由父母双方复合杂合突变遗传,第1处杂合变异来自父亲的UGT1A1基因第5号外显子1 456位T>G,导致编码蛋白的相对应氨基酸由酪氨酸变成天冬氨酸,合成的蛋白质产物结构异常,使UGT1A1 酶活性降低>95%[8]。第2处杂合变异为来自母亲的该基因第4号外显子1 091位C>T,为新近发现的突变位点,导致编码蛋白的第1 091位氨基酸由脯氨酸变成亮氨酸,导致合成的蛋白质产物结构异常。曾有文献报道UGT1A1 酶活性较野生型降低64.4%[9],目前研究报道较少。本例先证者为Tyr486Asp与Pro364Leu的复合杂合突变,相关报道较少,这种复合杂合突变可协同降低UGT1A1活性,作用高于单一突变[10]。基因检测显示该父母均为携带者,均无明显临床症状,但其子女患相关疾病风险为25%,表型正常的子女有2/3的风险为携带者,这为该家系评估再次妊娠风险、优生优育提供了重要参考。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
现代仪器与医疗(2022年1期)2022-04-19
中国生殖健康(2020年4期)2021-01-18
考试周刊(2017年26期)2017-12-12
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
湖北农业科学(2014年11期)2014-09-10
中学生英语高效课堂探究(2011年4期)2011-07-07
祝您健康(1985年6期)1985-12-30
