
微生物基因组精简优化的研究进展
2013-09-04 08:35王建莉王小元
生物工程学报 2013年8期
王建莉,王小元
1江南大学 食品科学与技术国家重点实验室,江苏 无锡 214122
2江南大学生物工程学院,江苏 无锡 214122
微生物基因组精简优化是构建合成生物学底盘细胞的重要策略[1]。在特定环境下,维持细胞正常代谢所需的最少基因群形成了该微生物的最小基因组[2]。获得最小基因组的方法主要有两种[3]:一种是“自上而下”,从现有微生物基因组中删除非必需基因;另一种是“自下而上”,用基本原料人工合成[4]。微生物基因组精简即采用“自上而下”法获得最小基因组。
微生物基因组精简一般选用遗传背景明晰的工业生产菌[5],如大肠杆菌、恶臭假单胞菌、枯草芽胞杆菌、谷氨酸棒杆菌、阿维链霉菌和酵母菌等。大肠杆菌的基因组精简比例已达38.9%[6]。精简区域的确定和精简策略的设计是微生物基因组精简的关键。文中针对这些关键问题进行分析讨论,为各种微生物基因组精简工作的开展提供参考。
1 微生物基因组必需基因
微生物必需基因是维持细胞生存所必需的,其突变通常具有致死性,因此对开发控制病原微生物的药物靶点具有重要意义[7]。掌握必需基因信息是基因组精简优化的前提条件。目前已经报道全基因组序列的微生物有4 325个,但其中必需基因信息被确定的只有20种 (表1)。微生物的必需基因一般通过培养基筛选确定。表1中除血链球菌Streptococcus sanguinisSK36和酿酒酵母Saccharomyces cerevisiae采用基本培养基筛选外,其他均采用丰富培养基。微生物中必需基因比例一般随着总基因数的增多而减少。生殖支原体Mycoplasma genitalium和流感嗜血杆菌Haemophilus influenzae的基因组较小,必需基因比例分别为 79%[8]和 40%[9];铜绿假单胞菌Pseudomonas aeruginosa基因组较大,其必需基因比例仅为5.6%[10]。多数微生物必需基因比例分布在10%~20%。用不同突变方法、筛选条件及判断标准得出的必需基因会有所差异。
必需基因在不同微生物中保守性很高。与亲缘关系较近的链球菌相比,血链球菌的必需基因编码的蛋白同源性为80%,而非必需基因编码的蛋白同源性为59%[11]。大肠杆菌的303个必需基因中有282个在肠杆菌中普遍存在[12],如在伤寒沙门氏菌Salmonella typhimurium、弗朗西斯菌Francisella novicida、不动杆菌Acinetobacter baylyiADP1和铜绿假单胞菌Pseudomonas aeruginosaPAO1 中分别存在 256 个[13]、126 个[14]、256个[15]和133个[16]。不动杆菌和铜绿假单胞菌有209个公共必需基因[10,15]。牙龈卟啉单胞菌Porphyromonas gingivalis有463个必需基因,其中364个基因与其他微生物必需基因有较高的同源性[17]。微生物基因组中不保守的必需基因往往与细胞独特的结构和功能有关。革兰氏阳性菌和革兰氏阴性菌的主要区别在于细胞膜壁结构和组成成分,二者基因组中与膜壁相关的必需基因也存在较大差异[11]。病原性微生物中的致病相关基因往往是必需基因,例如沙门氏菌中的O-抗原合成基因rfaL[13,18],弗朗西斯菌中也存在42个致病相关的必需基因[14];金黄色葡萄球菌Staphylococcus aureusN315和新月柄杆菌Caulobacter crescentus分别有51个[19]和48个[20]特有的必需基因。
微生物必需基因的保守性与其所编码蛋白在细胞中的作用密切相关。多数微生物的必需基因所编码的蛋白与DNA复制、细胞增殖、能量代谢、脂类合成等有关。血链球菌中DNA转
录、蛋白质合成或修饰、多糖代谢的相关基因中有50%~76%是必需基因[11]。幽门螺杆菌Helicobacter pylori中细胞分裂、蛋白合成、核酸代谢的相关基因中有25%~38%为必需基因[21]。谷氨酸棒杆菌Corynebacterium glutamicum中核酸代谢、细胞自身功能、胞内代谢反应的相关基因中也有20%~35%为必需基因[28]。大肠杆菌中核酸代谢和蛋白合成的相关基因中必需基因比例分别占28%和48%[24]。探索微生物必需基因的分布规律可以更好地设计细胞代谢途径[35],为构建合成生物学底盘细胞提供理论依据。

表1 各种微生物中必需基因数目及确定方法Table 1 Number of essential genes with methods to identify them in various microorganisms
确定微生物必需基因的常用方法主要有基因组序列比对法、全局转座插入突变法、单基因敲除法和反义RNA干扰法4种[24,36]。
1.1 基因组序列比对法
基因组序列比对法通过计算比对亲缘关系较远的微生物基因组确定必需基因[36]。微生物基因组信息的日益更新为基因组序列比对提供了有利条件。通过比对生殖支原体 (G+)和流感嗜血杆菌 (G−)的基因组序列,256个必需基因被确定[36-37]。通过比较8个较小微生物基因组,得到了仅含206个必需基因的最小基因组模型[38]。基因组序列比较的范围越广,得出的必需基因越少,但其保守性越强。通过比较3种大肠杆菌CFT073、DEL933和MG1655的基因组序列得出2 996个公共基因[39],而比较100个基因组则可得出63个保守基因[36]。基因组序列比对法可以在短时间内分析多种微生物基因的保守性,快速推测必需基因,但并不能准确确定某种微生物的全部必需基因。通过全基因组序列比对得出的最小基因组模型可能使微生物无法生存[40],但对基因组精简优化有很大参考价值。
1.2 全局转座插入突变法
全局转座插入突变法通过在染色体上高密度、高通量、超饱和地插入转座子使基因突变,同时结合基因组足迹法或基因组测序技术确定必需基因。转座子 (Transposon,Tn)可随机插入到基因组许多位点,转座子类型和转座子的插入密度、间隙、位置都会影响必需基因的确定。常用的转座子Tn5和Tn7被诱导修饰后其鉴定准确性可进一步提高[41]。在沙门氏菌中,通过阿拉伯糖诱导修饰获得转座子TnAraOut,进一步缺失必需基因使其表现出果聚糖依赖性[42]。在铜绿假单胞菌PAO1中,通过乳糖诱导修饰得到ISphoA/hah和ISlacZ/hah进而鉴定必需基因[10,16]。对转座子进行位点修饰可以在鉴定必需基因后进一步构建敲除突变株。在MG1655中,先采用Ⅰ-SceⅠ位点修饰的转座子 Tn-Kan-Ⅰ-SceⅠ鉴定必需基因,然后通过表达Ⅰ-SceⅠ酶获得了非必需基因的突变菌株库[43]。在弗朗西斯菌中,先采用FRT位点修饰的转座子ISFn2/FRT鉴定必需基因,进而表达Flp酶构建缺失突变株检验鉴定结果[14]。同时使用多种转座子鉴定微生物的必需基因可提高准确性,如在谷氨酸棒杆菌中应用IS31831和Tn5[28],在弗朗西斯菌中应用T17(ISFn1)、T18(ISFn2) 和 T20(ISFn2/FRT)[14]。在空肠弯曲菌Campylobacter jejuni中,使用不同的体外转座子Tn7和Mariner各得到625个和414个候选必需基因,其中233个共有基因被确定为必需基因[26];Stahl和Stintzi采用EZ-Tn5-Cm转座子确定出空肠弯曲菌195个必需基因[25],但与前者仅重叠26个[26]。通常情况转座子插入密度越大,间隙越小,插入位点越关键,结果越准确。全局转座插入突变法效率高,结果较为准确,应用较广泛,几乎所有的微生物都可以用全局转座插入法来确定其必需基因。但是,该方法也会错误地将不含转座子的小基因确定为必需基因[10],并容易将插入位置离3′端较近的基因[23]和同功酶编码基因[11]确定为非必需基因。
1.3 单基因敲除法
单基因敲除法通过逐一突变失活单个基因后考察菌株能否生存来确定必需基因[12]。非必需基因失活突变株会被筛选得到,但必需基因失活具有致死性。在大肠杆菌BW25113中,通过FRT/Flp位点特异性重组敲除获得了3 985个单突变株并进一步确定出303个必需基因[12]。在不动杆菌中通过同源重组敲除获得2 594个突变株,进而确定499个必需基因[15]。在酿酒酵母Saccharomyces cerevisiae中通过高通量敲除后考察突变株生长状况,进一步确定出1 105个必需基因[44]。不同方法确定的必需基因群会有差异。在大肠杆菌中,通过单基因敲除法[12]和通过全局转座插入突变法[24]确定的必需基因群仅重叠205个。这种差异主要是突变手段、生长条件或判断标准的不同所导致[12]。全局转座插入突变法可能将导致生长缓慢的突变基因鉴定为必需基因,而单基因敲除法严格按照菌株能否生存来确定必需基因[12]。通过将必需基因回补到mini-F质粒上使突变株表现回补依赖性也可以辅助鉴定必需基因,303个大肠杆菌必需基因中有35个并无回补依赖性[45],这主要是由筛选条件的不同所导致的。结合全基因组序列比对法可以减少单基因敲除的工作量。例如在血链球菌中,先通过全基因组序列比对得出较为保守的基因群,再对这些保守基因进行敲除来确定必需基因群[19]。虽然通过严格考察单个基因突变的致死性来确定必需基因很精确,但工作量很大,同时会错误地将同工酶基因鉴定为非必需基因,也会由于敲除效率低或筛选不当将非必需基因鉴定为必需基因。因此,该方法可以结合基因组序列比较法提高筛选通量,也可以结合基因回补方法提高鉴定准确率。
1.4 反义RNA干扰法
反义RNA干扰法先通过反义RNA阻止某特定基因的转录和翻译,再通过高效滴定法考察突变株的生长状况,将致死性的基因确定为必需基因[31]。该方法目前应用较少,只被应用于金黄色葡萄球菌中,在菌株WCUH29和RN4220中分别确定了150个[30]和658个[31]必需基因。这种方法快捷,在临床上有很好的应用前景,但目前应用有一定的局限性[30]。
2 微生物基因组精简策略
微生物基因组精简策略是构建合成生物学底盘细胞的关键所在,基因组精简主要基于同源重组 (Homologous recombination)、双链断裂修复 (Double strains break repair,DSBR)重组、位点特异性重组 (Site-specific recombination)、转座重组 (Transpositional recombination)及噬菌体转导 (Transduction)技术。其中,双链断裂修复重组和位点特异性重组均建立在同源重组的基础上,进一步实现基因敲除。转导技术通过噬菌体侵染细胞,可以将各菌株的大片段突变型整合入1个受体菌中,提高基因组精简效率。同源重组和双链断裂修复重组可实现无痕敲除,而位点特异性重组和转座重组会有碱基残留。转座重组一般可循环敲除获得基因组精简菌株,其余重组可通过重复敲除或转导获得基因组精简菌株。
2.1 基于同源重组的基因组精简
同源重组是指两个DNA分子之间同源区域交换实现序列重组的过程,并使得重组基因组稳定遗传。同源重组中的外源DNA包括环形质粒和线性片段两种。用于敲除的环形质粒主要是指自杀质粒,其敲除主要基于宿主菌自杀质粒的同源交换,而使用线性片段敲除主要基于Red重组的同源交换。通过同源重组进行基因组精简时往往需要同时引入抗性筛选标记 (Positive selection marker)和负筛选标记 (Negative selection marker)。
2.1.1 基于自杀质粒的同源重组
自杀质粒 (Suicide plasmid)通常在宿主菌中不能复制,可通过其携带的同源臂重组整合到染色体上敲除目的基因 (图1A)。质粒重组包括同源单交换和双交换:同源单交换在敲除过程中通过1个同源臂重组将整个质粒整合到基因组上,而同源双交换则通过2个同源臂重组替换目标基因片段。基因组精简时,可根据目标片段大小及重组效率选择合适的同源交换方式。在大肠杆菌中,自杀质粒同源双交换可以用于敲除中等长度片段[46]。在阿维链霉菌中,Komatsu等采用典型的自杀质粒同源双交换可一次性敲除1.48 Mbp的基因片段[47]。自杀质粒同源重组敲除虽可广泛地应用于多种微生物,但反复构建质粒较为繁琐,并需要较好地控制条件以减少假阳性转化子。自杀质粒同源双交换中第二轮交换效率较低,且容易回复突变为野生型表型,不利于敲除突变株的筛选。
2.1.2 基于线性DNA片段的同源重组
线性DNA片段是含有目的基因同源臂的敲除片段,通过电转化将敲除片段导入宿主菌中并与基因组发生同源重组,从而实现目的基因等位替换 (图1B)。线性DNA片段的同源重组最早应用于酿酒酵母中,通过在筛选标记两端添加40~50 bp的同源臂并PCR扩增获得敲除片段,将其电转入酵母中发生同源重组,进一步利用抗性标记筛选敲除突变株[48]。在酿酒酵母和栗酒裂殖酵母中,通过线性DNA片段同源重组已获得了一系列基因组精简菌株[49-50]。但在大肠杆菌中,RecBCD核酸外切酶会降解外源线性DNA片段,从而阻止敲除片段和基因组的同源重组[51]。Datsenko和Wanner利用λ-Red噬菌体中能抑制RecBCD的酶Gam、核酸外切酶Exo和单链结合蛋白Bet构建辅助质粒pKD46,提高了外源线性片段DNA的重组效率[52]和质粒同源重组效率[47]。线性片段同源重组在大肠杆菌的基因组精简中应用广泛,获得了许多基因组精简菌株,例如Δ16、Δ33[6]、MGF-01 和 DGF298[53]。通过λ-Red同源重组可敲除较长的基因片段,两轮同源重组后获得无痕敲除突变株[46,54],最后将不影响底盘细胞特性的突变基因通过P1转导[55]整合,获得最终基因组精简菌株[46,54]。在枯草芽胞杆菌中,Morimoto等结合线性DNA同源重组和自杀质粒两轮同源重组进行基因组精简 (图1C),前者用于敲除目的区域,后者用于去除筛选标记[56]。线性敲除片段的构建较自杀质粒更为简捷,重组效率也较高。
在同源重组中,往往通过引入合适的负筛选标记,并利用负筛选标记表达菌株对特定物质具有敏感性的特点,提高无痕敲除突变株的筛选效率。常用的负筛选标记有SacB、RpsL、Upp、Ura3或Ura4。其中,蔗糖致死性基因sacB存在于枯草芽胞杆菌,编码果聚糖蔗糖酶,在革兰氏阴性菌中表达sacB后可将蔗糖转化为果聚糖,堵塞在周质空间而使菌体死亡[57]。链霉素致死性基因rpsL编码的核糖体蛋白S12与链霉素形成复合物,增强了链霉素与细菌16S rRNA的结合能力从而阻止基因组转录[58]。5-氟尿嘧啶致死性基因upp编码尿嘧啶磷酸核糖转移酶,可将细胞外源的5-氟尿嘧啶转为细胞有毒物质抑制细胞生长[59]。5-氟乳清酸 (5-FOA)致死性基因pyrF或ura3和ura4编码乳清苷酸脱羧酶,可以催化5-氟乳清酸转化为有毒物质使细胞死亡[60]。在大肠杆菌中,常用SacB作为第二轮筛选标记,其筛选准确率平均达93%[61],敲除rpsL基因后,也可以选用RpsL作负筛选标记[6]。通过串联sacB和rpsL实现双重负筛选可进一步提高筛选准确率[46]。在谷氨酸棒杆菌中,引入sacB后通过质粒双轮同源交换也可以实现多轮无痕敲除[62]。在枯草芽胞杆菌中,通过先敲除或突变upp基因后再引入Upp作负筛选标记实现多轮敲除[56]。在酵母中,常用Ura3(或Ura4)作为基因组精简中的负筛选标记[49-50]。在栗酒裂殖酵母中,引入Ura4后可以进行多轮高效精确的无痕精简[50,63]。负筛选标记的引入有利于实现基因组的多轮无痕精简,提高同源重组的筛选效率。
2.2 基于DSBR的基因组精简
DSBR主要基于同源重组和基因组自修复机制实现基因敲除或基因组精简。Posfai等在1999年提出该方法[64],通过自杀质粒同源单交换在基因组中引入Ⅰ-SceⅠ识别位点S(18 bp),通过Ⅰ-SceⅠ酶表达从S点处中断基因组,从而诱导胞内SOS应急系统并启动基因组自身修复机制,主要在RecA核酸酶辅助下同源重组,从而删除筛选标记[64],达到无痕敲除。在大肠杆菌中,Kolisnychenko等通过DSBR重组进行目的片段(6~82 kb)的无痕敲除 (图1D)[65],Posfai等也采用该策略最终获得了一系列MDS(Multipledeletion series)精简菌株[66]。所构建的敲除片段含3段同源臂A、B和C,抗性筛选标记和2个Ⅰ-SceⅠ识别位点S,其中A和B为目的敲除区域的两端同源序列,C紧靠B为敲除区域末端的同源序列 (图1D)。先通过A和C同源重组替换目的片段,同时引入2个Ⅰ-SceⅠ识别位点S,进而通过B序列同源重组修复达到基因组无痕精简[65-66]。通过引入负筛选标记可提高DSBR敲除的筛选效率和准确性[67-68],Yu等在敲除盒中引入负筛选标记和1个S位点即可实现高效敲除(图1E)[68],他们利用不同启动子将Ⅰ-SceⅠ酶整合到pKD46上,构建两轮重组一体化质粒pRED1可进一步改善敲除系统,实现快速高效的敲除[68]。DSBR敲除系统也可以应用于恶臭假单胞菌[69]和谷氨酸棒杆菌[70]等微生物中。DSBR重组可以实现连续高效的无痕敲除,但往往需通过多次重叠PCR获得同源重组片段。
2.3 基于位点特异性重组的基因组精简
位点特异性重组通过特异性酶识别相应位点并删除位点间基因片段。位点特异性重组系统主要有Flp/FRT[52]和Cre/loxP[71]两种,它们常与同源重组结合应用于多种微生物的基因敲除。Flp和Cre是位点特异性识别酶,分别识别特异性位点FRT和loxP。FRT和loxP均含有2个反向的13 bp序列,以及1个8 bp的中心区域。特异性酶识别同向的特异性位点并重组去除位点之间的DNA序列。
Flp/FRT敲除系统多用于大肠杆菌、恶臭假单胞菌和酵母的基因敲除或基因组精简[72]。Cre/loxP较Flp/FRT系统应用更广泛,可应用于多种微生物的基因组精简。当敲除的目标片段较短时,只需合成1个敲除载体或片段,携带2个loxP位点,先通过同源重组替换目的基因,再通过Cre酶识别loxP位点重组去除筛选标记[73]。当敲除目标片段过长时,可分别构建两端基因的敲除载体或片段,各带1个loxP位点,先通过同源重组将loxP位点引入到目的区域两端,再通过Cre酶删除目的基因片段及筛选标记。在大肠杆菌中,Fukiya等通过敲除片段在目的区域两端各引入1个loxP位点 (图1F),可以敲除长达117 kb的片段[74],但其上游loxP位点位于抗性标记右侧,最终上游抗性标记未去除。在阿维链霉菌中,Komatsu等采用类似的原理,通过2个敲除载体分别引入loxP位点,可敲除1.51 Mbp的片段[47],其loxP均位于筛选标记和同源臂之间,最终可以去除筛选标记。Cre/loxP重组在每一轮敲除后会残留1个位点,干扰下一轮位点特异性识别。为了避免这种干扰,可设计携带突变序列的特异性位点,重组后序列发生突变,不再被Cre酶识别,进而可实现连续敲除。在谷氨酸棒杆菌中,Suzuki等通过设计带有突变位点的loxP-LE和loxP-RE建立Cre/lox突变系统 (图1G),可连续进行11轮敲除,每轮敲除的冗余片段长10~56 kb[75]。位点特异性重组较单一的同源重组敲除效率更高,无需引入负筛选标记,操作方便,广泛应用于多种微生物的基因敲除和基因组精简;但敲除后会残留特异性位点,不利于实现基因组无痕精简。
2.4 基于转座重组的基因组精简
基因转座子在转座酶的催化下可随机插入到基因组许多位点。复合式转座子不仅含转座酶(Transposase)基因,而且带有其他抗性基因或宿主基因,两端往往有重复序列。转座酶与转座子两端及DNA靶序列结合后将双链DNA交错切割,同时在转座子双链的两端切断不同单链,使产生的转座子游离端和靶序列游离端连接。利用复合式转座子的特点,可以将其应用于微生物基因组精简。在大肠杆菌中,Goryshin等利用Tn5转座子建立了以转座酶介导的基因组精简策略(图1H),最终成功精简基因组5.6%,获得菌株Δ20-4[76]。Tn5转座子由核心序列和两条倒置的IS50序列组成。核心序列中的转座基因表达转座酶Tnp-EK/LP,抗性基因供转座突变株筛选。IS50具有19 bp的倒置末端,外末端OE和内末端IE,其中OE介导整个转座子插入基因组,IE是转座酶Tnp-EK/LP的作用位点。当Tn5随机插入到大肠杆菌基因组上,表达转座酶Tnp-EK/LP结合到2个IE序列上,从该处单链切断使其产生粘性末端,同时删除核心序列。此时,有活性的转座酶Tnp-EK/LP使得2个IE序列在基因组内部转位,并使插入位置的DNA形成2个游离端。当左边IE的粘性末端与右边的游离端连接时实现基因片段 (4~23 kb)敲除 (图1H),而右边IE与左边游离端连接形成回补质粒[76]。基于转座酶重组的基因组精简可以筛选得到较多敲除突变株,效率较高,同时可以进行基因功能研究;但会在基因组上残留若干对端部序列,且随机性过强,不易准确控制精简区域,还需通过全基因组测序来鉴定敲除区域。
2.5 基于多种敲除系统复合的基因组精简
利用各敲除系统的典型特点,综合多种敲除系统可建立更为理想和高效的基因组精简策略。在谷氨酸棒杆菌中,Suzuki等结合Cre/loxP和DSBR敲除系统实现了连续无痕敲除 (图2A)[70]。构建两个自杀质粒,通过其同源双交换在目的区域的两端同时引入loxP位点和Ⅰ-SceⅠ识别位点S。其中loxP位点处于抗性筛选标记和负筛选标记间,而S位点处于同源臂和负筛选标记之间。先通过Cre/loxP位点特异性重组删除目的片段,再通过DSBR重组去除筛选标记及loxP位点,获得无痕敲除菌株[70]。Cre/loxP和DSBR敲除系统的有序结合实现了高效的连续无痕精简。
在恶臭假单胞菌中,Leprince等通过结合转座重组和位点特异性重组建立了一套基于Tn5和FRT/Flp的基因组精简策略 (图2B)[72]。通过对Tn5衍生的小型转座子进行单个FRT位点修饰,形成两种带有不同筛选标记的小型转座子Mini-Tn5KpF和Mini-Tn5TF,分别含有1个OE和IE,FRT位点紧靠OE序列。前者使突变株具有Km抗性和基因pyrF引起的5-FOA敏感性,后者使菌株具有亚碲酸钾 (Tel)抗性。首先通过Mini-Tn5KpF的随机插入到恶臭假单胞菌基因组中获得单插入突变株库SMT(63株)。再从中随机选择9株,插入Mini-Tn5TF转座子,获得双插入突变株库TMT(573株)。通过测序确定TMT菌株的突变情况,筛选合适的突变株 (7株),其FRT位点同向,Mini-Tn5KpF和Mini-Tn5TF分别在所敲除区域上游和下游。通过Flp酶识别FRT位点重组,去除非必需片段和抗性筛选标记,获得精简菌株 91-Δ1和 407-Δ1[72]。在Δ1菌株中同时插入 Mini-Tn5KpF和Mini-Tn5TF获得Δ1SMT突变株库,进一步通过三亲杂交整合两种转座的突变型,获得多种Δ1TMT菌株。Δ1TMT包括两种转座子,3个FRT位点,表达Flp酶后有多种重组情况 (图2B),均可以继续精简获得91-Δ2和 407-Δ2,精简比例最高为7.4%[72]。该敲除策略可以快速实现较长区域的敲除,所构建的突变株库可以进行基因功能研究;但该策略随机性强,最终在基因组上残留FRT位点,甚至残留抗性标记和转座子序列。
在大肠杆菌中,Yu等通过结合转座重组、位点特异性重组及P1噬菌体转导实现了基因组精简 (图 2C)[77]。通过对Tn5转座子进行单个loxP位点修饰得到TnKloxP(Kanr)和TnCloxP(Cmr)转座子,分别在基因组中随机插入构建突变株库,并确定各突变株插入位置。根据目的敲除区域,从两种突变株库中分别选择相应的突变株,其loxP位点同向并分别位于目的敲除区域两端。先通过P1转导整合突变基因,再诱导Cre酶识别loxP位点删除目的片段 (59~117 kb)。重复进行转导和位点特异性重组可实现连续敲除,最终获得精简菌株 CDΔ3456,精简比例达6.7%[77]。这种敲除策略通过转座子高通量插入可以获得较多突变株,结合P1转导和Cre/loxP可以快速实现目的片段的敲除;但该策略有一定的随机性,需通过基因组测序鉴定转座突变基因,也会在基因组上残留抗性标记和loxP位点。
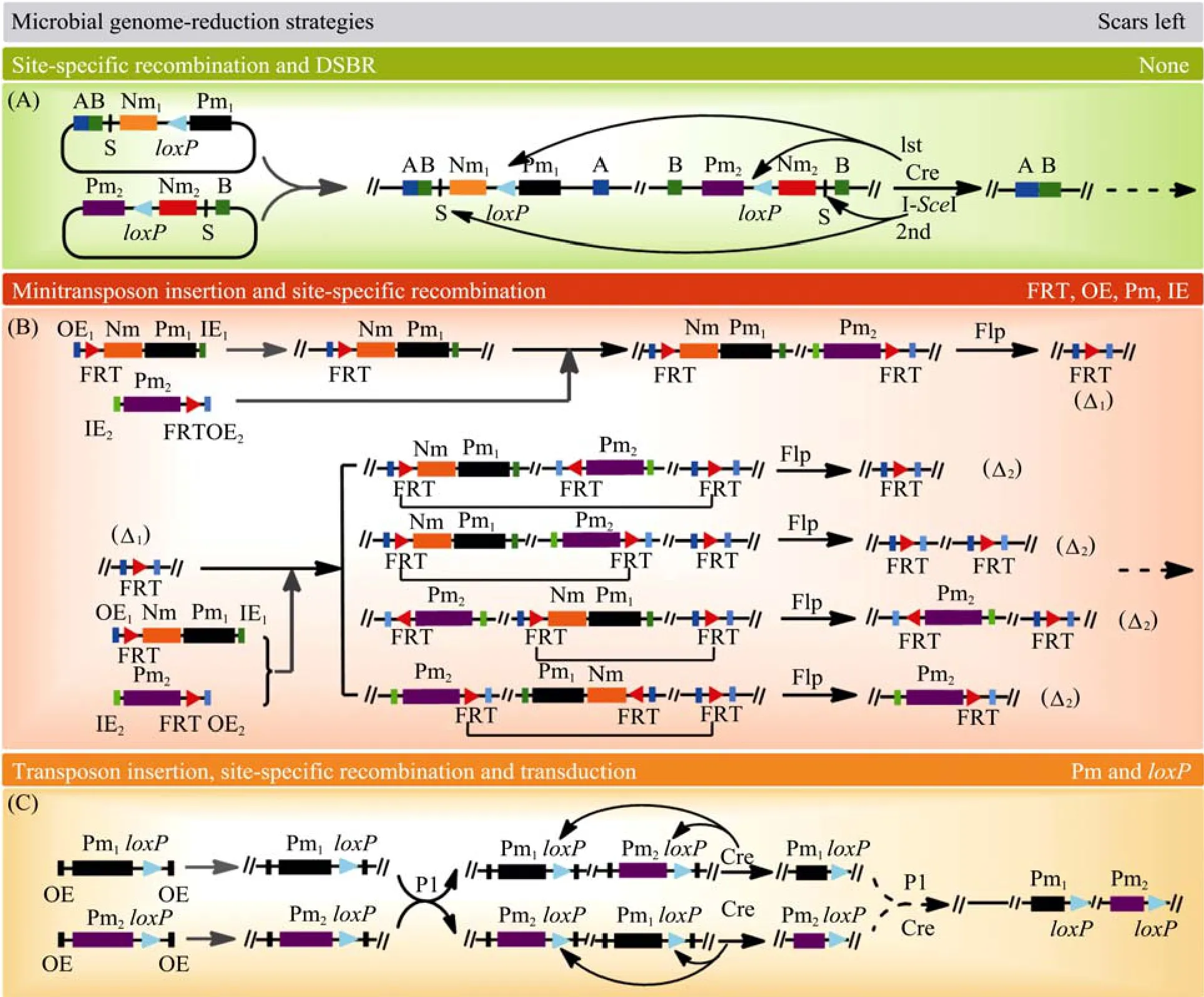
图2 基于多种重组系统复合的微生物基因组精简策略Fig.2 Microbial genome-reduction strategies by combining different recombination systems.(A)The combined strategies of the Cre/loxP system and DSBR.S and loxP sites are integrated into the chromosomes via homologous recombination of the plasmids[70].(B)The combined strategies of the transposon insertion and the FLP/FRT site-specific recombination.Two rounds of the deletions are shown[72].(C)The combined strategies of the Tn5 transposon,Cre/loxP excision system and P1 transduction.After insertions of two modified Tn5,the two mutant strains with a loxP site are combined into a single strain by P1 transduction[77].The target DNA between the two loxP sites in the new mutant strain is deleted by Cre recombination.The double oblique hyphens represent the break regions of the chromosomal DNA.The letters A and B represent the homologous arms of the target DNA on the chromosome.OE and IE represent external and internal transposon ends in Tn5,respectively.Pm and Nm represent positive and negative selection markers,respectively.
3 微生物基因组精简现状
通过基因组精简优化,微生物细胞的生产潜力和可控性会大幅度增强[78],可以更有效地合成目的产物[79]。目前基因组精简优化的大肠杆菌、恶臭假单胞杆菌、枯草芽胞杆菌、谷氨酸棒杆菌、链霉菌及酵母菌均有报道 (表2)。其中,大肠杆菌基因组精简幅度最高,为38.9%,枯草芽胞杆菌和阿维链霉菌的基因组精简为18%~23%,其他微生物基因组精简低于10%。
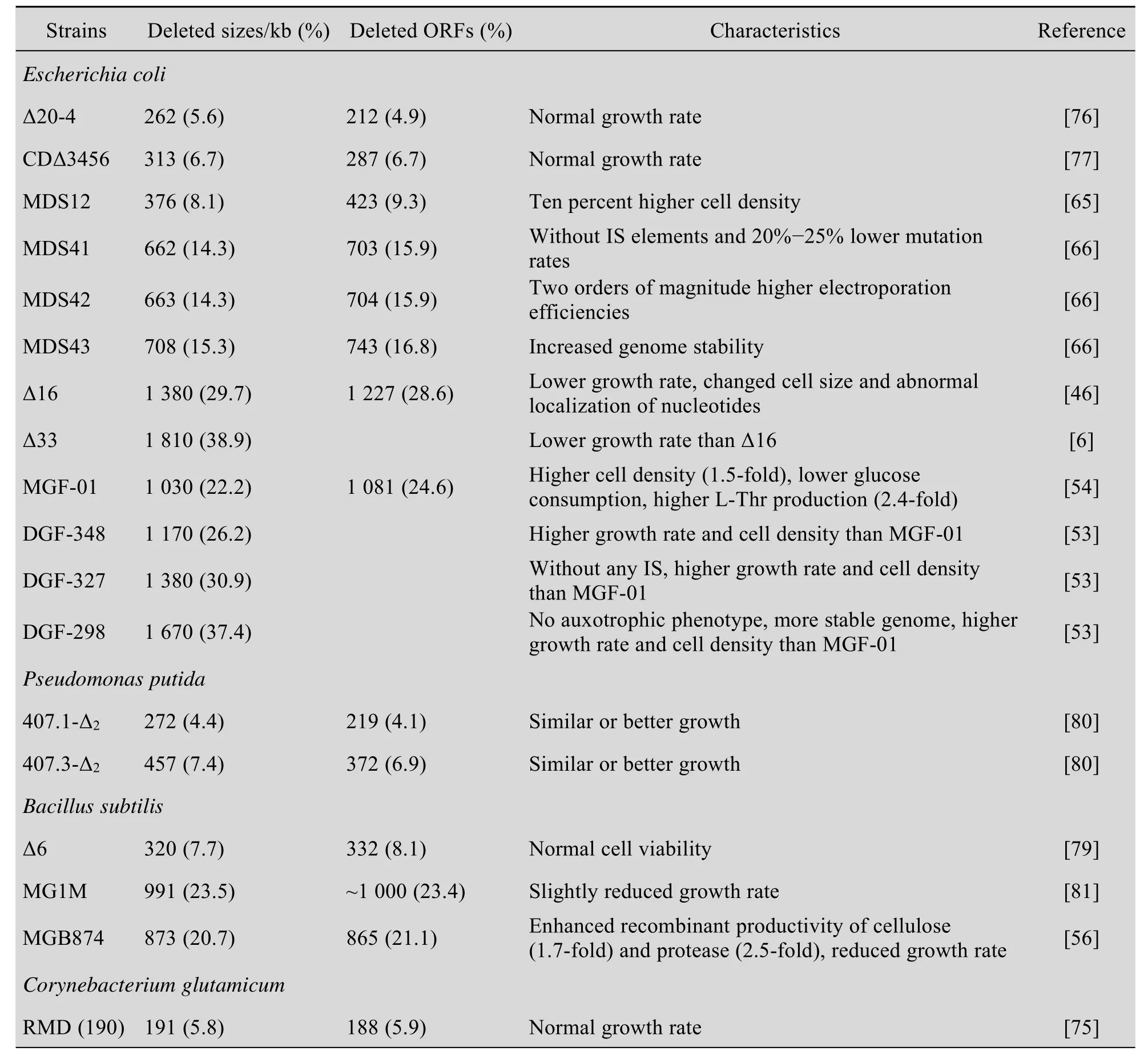
表2 不同微生物的精简菌株特性Table 2 Characteristics of different microorganisms with minimal genome
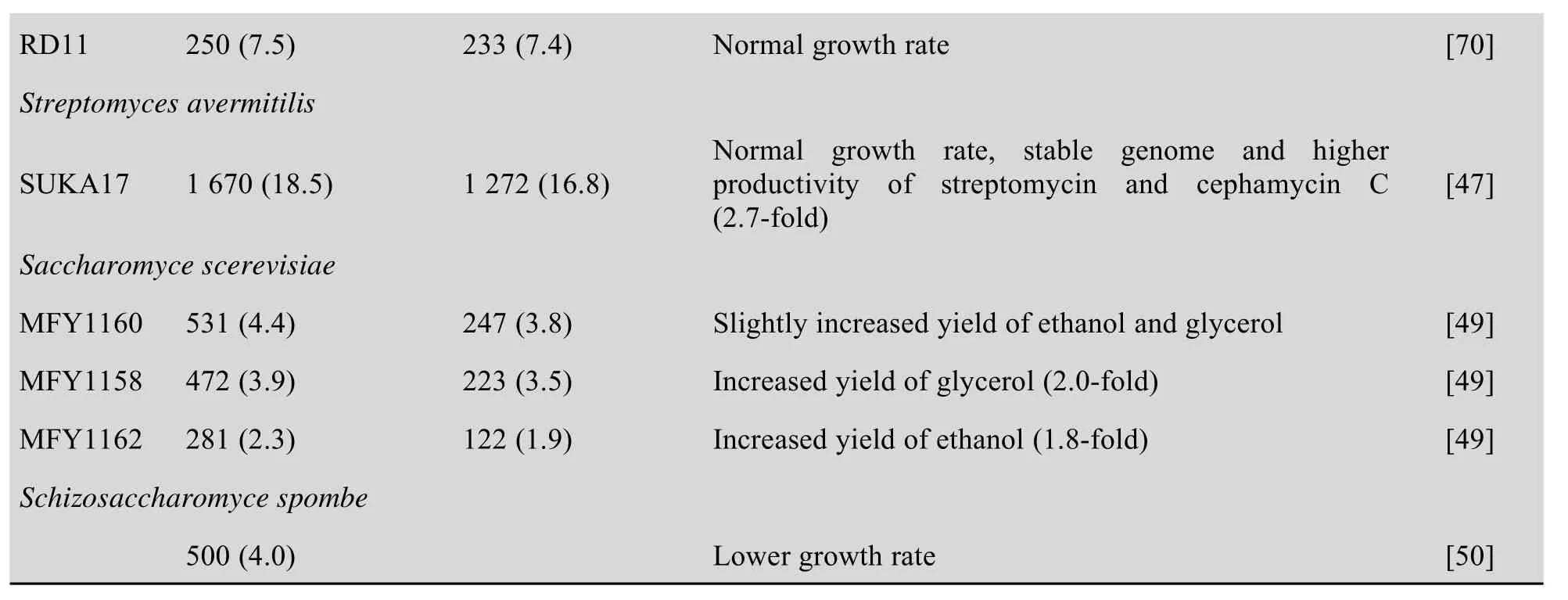
续表2
3.1 大肠杆菌
大肠杆菌遗传背景清晰,分子操作技术成熟,工业应用潜力大,是构建合成生物学底盘细胞的最佳选择之一。自2002年以来,大肠杆菌基因组精简幅度从5.6%提高至38.9%,其中,MGF(Minimumgenomefactory)和DGF(Designed genome factory)系列精简菌株由W3110改造,其余精简菌株则来自MG1655。这些精简菌株生长基本正常,多数表现出底盘细胞的优良特性。
精简菌株Δ20-4[76]和 CDΔ3456[77]均通过随机精简非必需基因而获得,生长正常,但未表现出其他优良特性。Kolisnychenko等通过DSBR敲除系统定性敲除11个较大的和6个较小的K-组岛 (K-island)获得精简菌株MDS12[65],其在基本培养基中生长正常,稳定期细胞量提高了约10%[65]。Posfai等在 MDS12的基础上,通过DSBR敲除系统进一步精简了含基因组岛的IS序列和含IS序列的单个基因,获得 MDS41、MDS42和MDS43,其生长良好、遗传稳定、电转化效率提高[66]。通过转录组学分析发现,MDS42中L-苏氨酸 (L-Thr)合成相关基因转录水平有较大变化,其重组菌较野生菌产量提高83%[82]。Csorgo等通过突变SOS调控蛋白RecA和LexA以及SOS诱导的DNA聚合酶PolII、PolIV和PolV,提高了MDS42的遗传稳定性[83]。这3个DNA聚合酶编码基因的敲除突变株MDS42pdu的随机突变率降低了50%左右,但生长依然正常[83]。MDS42pdu在施加环境胁迫或表达毒力蛋白 (ORF238)情况下,仍具有较强的遗传稳定性,可应用于毒力蛋白的过表达研究[83]。
Hashimoto等参考 PEC(Profiling ofE.coliChromosome,http://shigen.lab.nig.ac.jp/ecoli/pec/)的非必需基因信息,通过同源重组和P1转导获得菌株Δ16,精简比例高达29.7%[46]。和MDS43相比,Δ16所精简的单个大片段覆盖范围更宽,总敲除基因数更多,两者公共敲除基因有435个[54];但是Δ16生长减慢,细胞更长更宽,胞内核酸分布更分散、更贴壁[46]。Iwadate等在Δ16的基础上,通过同源重组进一步精简获得Δ33,其精简比例为38.9%,是目前最高的,但其生长特性较差[6]。Δ16和Δ33特性异常,并不适合作为底盘细胞。因此,理性选择精简区域对基因组精简优化非常关键。日本MGF项目团队在CGH分析的基础上,以3 Mbp的大肠杆菌小基因组模型为精简目标[78,84]。Mizoguchi等参考PEC信息通过同源重组和P1转导理性敲除大片段获得菌株MGF-01,精简比例为 22.2%[54]。MGF-01所精简的单个大片段覆盖范围较Δ16窄,所敲除基因总数比Δ16少146个,比MDS43多298个[54];MGF-01与Δ16所敲除基因重叠 493个,与MDS43重叠568个[54]。MGF-01比野生型生长更好,其L-Thr代谢工程菌的产量更高,并具有较高的底物转化率[54]。在生长和L-Thr生产方面,MGF-01比 MDS43和Δ16更有优势。通过对MGF-01进行转录组学分析,发现受YdfH抑制的饥饿感应蛋白编码基因rpsA上调[85],因此,YdfH的缺失可能是导致其生长加快的原因之一。Hirokawa等在MGF-01的基础上,进一步敲除2~3个大片段区域获得 MGF-02,其生长较MGF-01更好[53]。在这2个MGF菌株基础上,Hirokawa等通过回补表达ilvG和rph基因后,继续合理精简基因组,最终构建了一系列DGF菌株,精简比例最高达37.4%[53]。其中,DGF-348具有明显的生长优势,DGF-327和DGF-298不含任何IS序列,生长更快且细胞终浓度增加,无营养缺陷性,DGF-298还表现出更好的遗传稳定性[53]。通过对DGF各精简菌株进行转录组水平分析,发现部分热激性蛋白和蛋白激酶的编码基因转录水平下调,这可能是其生长速率提高的主要原因[53]。
3.2 其他菌株
除大肠杆菌外,目前已报道的基因组精简优化的原核微生物主要有恶臭假单胞菌、枯草芽胞杆菌、谷氨酸棒杆菌和阿维链霉菌,真核微生物主要是酿酒酵母和栗酒裂殖酵母。
恶臭假单胞菌主要用于生产新型材料聚羟基脂肪酸 (Polyhydroxyalkanoates,PHA),在合成塑料、燃料、高附加值医疗产品等方面有很高的工业应用价值。Leprince等通过转座重组和FRT/Flp位点特异性重组随机敲除以基因组岛序列为主的非必需基因[72,80],获得菌株407.1-Δ2和407.3-Δ2;与野生型相比,这些菌株的生长正常或更好[80]。菌株407.3-Δ2缺失117个假蛋白和7个转座酶的编码基因,与遗传性更稳定的大肠杆菌MDS42的敲除基因重叠较多,因此推测其遗传性更稳定[80]。枯草芽胞杆菌也是重要的工业生产菌,主要用于各种分泌蛋白酶的生产,近年来作为典型的模式微生物被广泛研究,其精简菌株主要有Δ6、MG1M和MGB874。Westers等通过敲除Prophage区域及聚酮化合物合成酶操纵子pks获得Δ6,其生长代谢正常,但运动性和某些蛋白的分泌受到影响[79]。Ara等通过定性精简Phage系列基因和抗生素合成相关基因获得MG1M,其生长正常,纤维素酶生产能力提高[81]。Morimoto等通过同源重组敲除多个Prophage区域和11个非必需基因簇获得MGB874,其重组菌纤维素酶和蛋白激酶的产量分别提高1.7倍和2.5倍,底物利用率也有所提高[56];但MGB874具有生长缺陷性[56],有待进一步改善。谷氨酸棒杆菌常用于氨基酸、核苷酸和有机酸的生产。Suzuki等通过Cre/lox突变型位点特异性重组获得菌株 RMD(190)[75]和 RD11[70]。RMD(190)所精简的区域主要是11个菌株特异性基因组岛(Strain-specific islands),RD11所精简区域除基因组岛外还有功能未知的冗余基因,二者均生长正常[70]。阿维链霉菌是重要的驱虫菌素等次级代谢物质生产菌,有较强的初级代谢能力,可提供充足的前体和能量[47]。Komatsu等通过结合同源重组和Cre/loxP位点特异性重组获得菌株SUKA17。SUKA17中精简了基因组中78%的转座序列,总基因组精简比例为18.5%[47]。SUKA17生长正常,遗传稳定,并具有良好的链霉素和头霉素C生产特性,其中头霉素C的合成量约提高 2.7 倍[47]。
酿酒酵母主要用于生产酒精、甘油等。Murakami等通过计算预测了609个目的敲除基因,采用PCR介导的基因组断裂重组获得了一系列精简菌株[49]。其中,菌株MFY1160基因组精简比例最高 (4.4%),但其酒精和甘油的产量没有明显提高,MFY1158的甘油产量提高2倍,MFY1162酒精产量提高1.8倍[49]。栗酒裂殖酵母主要用于蛋白酶的生产。通过同源重组获得栗酒裂殖酵母精简菌株,精简区域多于500 kb,但菌体有一定的生长缺陷性[50]。栗酒裂殖酵母的蛋白激酶缺失菌株也可大幅度提高外源蛋白的表达量,蛋白激酶的缺失可以减少对外源蛋白的降解,有利于构建高效的外源蛋白生产菌。例如,利用缺失7个蛋白激酶的菌株MGF341构建的重组菌生产人体生长激素,产量可提高30倍[50,63]。
4 展望
多种微生物通过基因组精简优化后表现出优良特性,可见基因组精简优化是构建合成生物学底盘细胞的有效手段。基因组精简优化的前提是确定精简靶区域,其关键是建立完善的大片段敲除策略。目前仍有多种微生物存在较多功能未知基因,基因组精简策略尚不成熟,这些问题是基因组精简优化工作的主要瓶颈。
首先,理性设计敲除靶区域和定性精简对定向优化基因组至关重要。基因组精简优化并非盲目敲除非必需基因,而是尽可能改造出只包含有利基因的微生物。同时,根据目的产物或功能模块可进一步优化基因组以提高底盘细胞的适配性。每个必需基因的缺失都有可能导致生长缺陷性,应尽量避免敲除这些基因。通过代谢网络计算进行定性精简可获得具有优良特性的菌株,现有的小基因组微生物也为精简区域的选择提供了重要参考。大肠杆菌精简菌株的敲除靶区域多为基因组岛序列、Prophage区域、IS序列和冗余基因,还包括较多膜壁结构相关基因。膜壁附属结构及膜蛋白相关基因的敲除有利于电转化效率的提高[66]。革兰氏阴性菌膜壁结构包括鞭毛、纤毛、脂多糖、荚膜多糖和胞外分泌物等,其非必需结构的精简可能会减少菌体的糖耗,有利于提高细胞“经济效益”,改善细胞生产特性。恶臭假单胞菌天冬氨酸转氨酶突变株的脂多糖合成量提高,而PHA合成量降低[86],说明脂多糖的过量合成可能不利于PHA的合成[87-88]。对大肠杆菌脂多糖的结构进行精简优化[89],可提高细胞壁通透性,有利于产物分泌。
其次,建立精确高效的精简策略是优化基因组的关键所在。基于同源重组、DSBR重组和位点特异性重组的精简策略可以精确敲除目的基因片段,有助于实现基因组的定性精简。目前,多数微生物的大片段敲除技术尚不成熟或有待改善,可以根据不同微生物的遗传特性选择合适的敲除策略,并从菌株特性和具体策略两方面进一步提高精简效率。通过构建相应的Red重组辅助质粒,敲除内源核酸酶RecJ、ExoI、ExoVII、ExoX和λExo,可以提高同源重组效率[90],进一步改造膜壁结构可以改善转化效率。综合不同敲除系统的优势构建更高效的精简策略。例如,结合DSBR和Cre/loxP重组实现高效连续的无痕精简;将Red酶系敲入到基因组上代替辅助质粒简化大片段敲除步骤;将不同重组酶 (如Red、Cre、Ⅰ-SceⅠ等)整合到1个质粒上也可以简化大片段敲除步骤。当相邻的敲除目的片段间只有几个必需基因时,可以合理设计敲除盒实现相邻目的片段的一步敲除。例如,采用DSBR敲除系统时,将必需基因连在敲除盒的同源臂A和B之间(图1E);采用Cre/loxP敲除系统时,将必需基因连在敲除盒的同源臂与loxP位点之间 (图1F和G)。此外,涉及转座重组的敲除策略具有一定的随机性,但通过高通量转座插入可以获得较多突变株,进而快速实现非必需基因敲除,同时便于功能未知基因的研究。在较多遗传背景不清晰的微生物中,应用转座重组可能会推动基因组精简优化工作的开展。
最后,深入了解微生物的各种调控机制是进一步优化精简菌株的必要条件。微生物是一个全局精密调控的有机整体,通过控制微生物调控因子的表达,可以更好地优化精简菌株的生理特性。例如RpoS是影响生长的关键调控因子之一,对其进行位点突变可能会提高菌体生长速率。表达某些关键基因也可能改善菌体生长特性,同时避免高盐环境和过量的ppGpp,或减少DksA、H-NS、Lrp蛋白和毒力蛋白的表达,可以降低微生物的生长抑制性[91-92]。通过增加基因组的起始复制子ori个数也可能会提高细胞生长速率或蛋白表达量[93],进一步改造相关基因还可以提高胞内代谢能量和还原力。此外,异源表达较长基因簇的技术日益成熟,包括化学合成基因簇[94]和基因组敲入整合[95],也为基因组精简优化菌株开辟了更广阔的应用前景。
基因组精简优化使微生物的整个系统更具潜力和操作性,同时也使我们更好地认识和利用微生物。具有优良特性的小基因组微生物作为合成生物学底盘细胞为功能模块的引入提供了基础平台,选择适配性较好的底盘细胞可使模块高效发挥作用,最终成为理想的“细胞工厂”。这些“细胞工厂”在代谢产物生产、环境废弃物降解、细胞毒素检测和生物能源开发等领域具有更广阔的应用前景。
[1]Diez MS, Lam CM, Leprince A, et al.(Re-)construction,characterization and modeling of systems for syntheticbiology.Biotechnol J,2009,4(10):1382−1391.
[2]Mushegian A.The minimal genome concept.Curr Opin Genet Dev,1999,9(6):709−714.
[3]Luisi PL.Toward the engineering of minimal living cells.Anat Rec,2002,268(3):208−214.
[4]Forster AC,Church GM.Synthetic biology projectsin vitro.Genome Res,2007,17(1):1−6.
[5]Choffnes ER,Relman DA,Pray L,et al.The Science and Applications of Synthetic and Systems Biology:Workshop Summary.Washington(DC):National Academies Press(US),2011.
[6]Iwadate Y,Honda H,Sato H,et al.Oxidative stress sensitivity of engineeredEscherichia colicells with a reduced genome.FEMS Microbiol Lett,2011,322(1):25−33.
[7]Juhas M,Eberl L,Church GM.Essential genes as antimicrobial targets and cornerstones of synthetic biology.Trends Biotechnol,2012,30(11):601−607.
[8]Glass JI,Assad-Garcia N,Alperovich N,et al.Essential genes of a minimal bacterium.Proc Natl Acad Sci USA,2006,103(2):425−430.
[9]Akerley BJ,Rubin EJ,Novick VL,et al.A genome-scale analysis for identification of genes required for growth or survival ofHaemophilus influenzae.Proc Natl Acad Sci USA,2002,99(2):966−971.
[10]Liberati NT,Urbach JM,Miyata S,et al.An ordered,nonredundant library ofPseudomonas aeruginosastrain PA14 transposon insertion mutants.Proc Natl Acad Sci USA,2006,103(8):2833−2838.
[11]Xu P,Ge X,Chen L,et al.Genome-wide essential gene identification inStreptococcus sanguinis.Sci Rep,2011,1:125.
[12]Baba T,Ara T,Hasegawa M,et al.Construction ofEscherichiacoliK-12 in-frame, single-gene knockout mutants:the Keio collection.Mol Syst Biol,2006,2(1):2006 0008.
[13]LangridgeGC,Phan MD,TurnerDJ,etal.Simultaneous assay of everySalmonella typhigene using one million transposon mutants.Genome Res,2009,19(12):2308−2316.
[14]Gallagher LA,Ramage E,Jacobs MA,et al.A comprehensive transposon mutant library ofFrancisella novicida,a bioweapon surrogate.Proc Natl Acad Sci USA,2007,104(3):1009−1014.
[15]de Berardinis V,Vallenet D,Castelli V,et al.A complete collection of single-gene deletion mutants ofAcinetobacter baylyiADP1.Mol Syst Biol,2008,4(1):174.
[16]Jacobs MA,Alwood A,Thaipisuttikul I,et al.Comprehensive transposon mutant library ofPseudomonas aeruginosa.Proc Natl Acad Sci USA,2003,100(24):14339−14344.
[17]Klein BA,Tenorio EL,LazinskiDW,etal.Identification of essential genes of the periodontal pathogenPorphyromonasgingivalis. BMC Genomics,2012,13(1):578.
[18]Knuth K,Niesalla H,Hueck CJ,et al.Large-scale identification of essentialSalmonellagenes by trapping lethal insertions.Mol Microbiol,2004,51(6):1729−1744.
[19]Soo K,Lee JY,Song JH,et al.Screening of essential genes inStaphylococcus aureusN315 using comparative genomics and allelic replacement mutagenesis.J Microbiol Biotechnol,2006,16(4):623−632.
[20]Christen B,Abeliuk E,Collier JM,et al.The essential genome of a bacterium.Mol Syst Biol,2011,7(1):528.
[21]Salama NR,Shepherd B,Falkow S.Global transposon mutagenesis and essential gene analysis ofHelicobacter pylori.J Bacteriol,2004,186(23):7926−7935.
[22]Hutchison Iii CA.Global transposon mutagenesis and a minimalMycoplasmagenome.Science,1999,286(5447):2165−2169.
[23]French CT,Lao P,Loraine AE,et al.Large-scale transposon mutagenesis ofMycoplasma pulmonis.Mol Microbiol,2008,69(1):67−76.
[24]Gerdes SY,Scholle MD,Campbell JW,et al.Experimental determination and system level analysis of essential genes inEscherichia coliMG1655.J Bacteriol,2003,185(19):5673−5684.
[25]Stahl M,Stintzi A.Identification of essential genes inC.jejunigenome highlights hyper-variable plasticity regions.Funct Integr Genomics,2011,11(2):241−257.
[26]Metris A,Reuter M,Gaskin DJ,et al.In vivo and in silico determination of essential genes ofCampylobacterjejuni.BMC Genomics,2011,12(1):535.
[27]Kobayashi K,Ehrlich SD,Albertini A,et al.EssentialBacillus subtilisgenes.Proc Natl Acad Sci USA,2003,100(8):4678−4683.
[28]Suzuki N, Okai N, Nonaka H, et al.High-throughput transposon mutagenesis ofCorynebacterium glutamicumand construction of a single-gene disruptant mutant library.Appl Environ Microbiol,2006,72(5):3750−3755.
[29]Thanassi JA,Hartman-Neumann SL,Dougherty TJ,et al.Identification of 113 conserved essential genes using a high-throughput gene disruption system inStreptococcuspneumoniae.Nucleic Acids Res,2002,30(14):3152−3162.
[30]Ji Y,Zhang B,Van SF,et al.Identification of criticalstaphylococcalgenesusing conditional phenotypes generated by antisense RNA.Science,2001,293(5538):2266−2269.
[31]Forsyth RA,Haselbeck RJ,Ohlsen KL,et al.A genome-wide strategy for the identification of essential genes inStaphylococcus aureus.Mol Microbiol,2002,43(6):1387−1400.
[32]ChaudhuriRR,Allen AG,Owen PJ,etal.Comprehensive identification of essentialStaphylococcus aureusgenes using Transposon-Mediated Differential Hybridisation (TMDH).BMC genomics,2009,10(1):291.
[33]Sassetti CM,Boyd DH,Rubin EJ.Genes required formycobacterialgrowth defined by high density mutagenesis.Mol Microbiol,2003,48(1):77−84.
[34]Giaever G,Chu AM,Ni L,et al.Functional profiling of theSaccharomyces cerevisiaegenome.Nature,2002,418(6896):387−391.
[35]Gerdes S,Edwards R,Kubal M,et al.Essential genes on metabolic maps.Curr Opin Biotechnol,2006,17(5):448−456.
[36]Koonin EV.Comparative genomics,minimal gene-sets and the last universal common ancestor.Nat Rev Microbiol,2003,1(2):127−136.
[37]Mushegian AR,Koonin EV.A minimal gene set for cellular life derived by comparison of complete bacterial genomes.Proc Natl Acad Sci USA,1996,93(19):10268−10273.
[38]Gil R,Silva FJ,Pereto J,et al.Determination of the core of a minimal bacterial gene set.Microbiol Mol Biol Rev,2004,68(3):518−537.
[39]Welch RA,Burland V,Plunkett G,et al.Extensive mosaic structure revealed by the complete genome sequence of uropathogenicEscherichia coli.Proc Natl Acad Sci USA,2002,99(26):17020−17024.
[40]Riley M,Serres MH.Interim report on genomics ofEscherichia coli.Annu Rev Microbiol,2000,54(1):341−411.
[41]Judson N, Mekalanos JJ. TnAraOut, a transposon-based approach to identify and characterize essential bacterial genes. Nat Biotechnol,2000,18(7):740−745.
[42]Kim JN,Youm GW,Kwon YM.Essential genes inSalmonella enteritidisas identified by TnAraOut mutagenesis.Curr Microbiol,2008,57(4):391−394.
[43]Kang Y,Durfee T,Glasner JD,et al.Systematic mutagenesis of theEscherichia coligenome.J Bacteriol,2004,186(15):4921−4930.
[44]Winzeler EA.Functional Characterization of theS.erevisiaegenome by gene deletion and parallel analysis.Science,1999,285(5429):901-906.
[45]Kato J,Hashimoto M.Construction of consecutive deletions of theEscherichia colichromosome.Mol Syst Biol,2007,3(1):132.
[46]Hashimoto M,Ichimura T,Mizoguchi H,et al.Cell size and nucleoid organization ofengineeredEscherichia colicells with a reduced genome.Mol Microbiol,2005,55(1):137−149.
[47]Komatsu M,Uchiyama T,Omura S,etal.Genome-minimizedStreptomyceshostfor the heterologous expression of secondary metabolism.Proc NatlAcad Sci USA,2010,107(6):2646−2651.
[48]Baudin A,Ozier-Kalogeropoulos O,Denouel A,et al.A simple and efficient method for direct gene deletion inSaccharomycescerevisiae.Nucleic Acids Res,1993,21(14):3329−3330.
[49]Murakami K,Tao E,Ito Y,et al.Large scale deletions in theSaccharomyces cerevisiaegenome create strains with altered regulation of carbon metabolism.ApplMicrobiolBiotechnol,2007,75(3):589−597.
[50]Giga-Hama Y,TohdaH,TakegawaK,etal.Schizosaccharomycespombeminimum genome factory.Biotechnol Appl Biochem,2007,46(3):147−155.
[51]Lorenz MG,WackernagelW.Bacterialgene transfer by natural genetic transformation in the environment.Microbiol Rev,1994,58(3):563−602.
[52]Datsenko KA,Wanner BL.One-step inactivation of chromosomal genes inEscherichia coliK-12 using PCR products.Proc Natl Acad Sci USA,2000,97(12):6640−6645.
[53]Hirokawa Y,Kawano H,Tanaka-Masuda K,et al.Genetic manipulations restored the growth fitness ofreduced-genomeEscherichiacoli.JBiosci Bioeng,2013.
[54]Mizoguchi H, Sawano Y, Kato J, et al.Superpositioning of deletions promotes growth ofEscherichia coliwith a reduced genome.DNA Res,2008,15(5):277−284.
[55]Litz-Jackson S,Miller AH,Burgess GS,et al.Dissociation of nuclear events on p21 RAS transformation of FDC-P1 myeloid cells:c-jun/AP-1 expression versus c-myc transcription.Blood,1992,79(9):2404−2414.
[56]Morimoto T,Kadoya R,Endo K,et al.Enhanced recombinant protein productivity by genome reduction inBacillus subtilis.DNA Res,2008,15(2):73−81.
[57]Steinmetz M,Le Coq D,Djemia HB,et al.Genetic analysis ofsacB,the structural gene of a secreted enzyme,levansucrase ofBacillus subtilisMarburg.Mol Gen Genet,1983,191(1):138−144.
[58]Cole ST,Brosch R,Parkhill J,et al.Deciphering the biology ofMycobacterium tuberculosisfrom the complete genome sequence.Nature,1998,393(6685):537−544.
[59]Tu AH,Turnbough CL.Regulation ofuppexpression inEscherichia coliby UTP-sensitive selection of transcriptional start sites coupled with UTP-dependent reiterative transcription. J Bacteriol,1997,179(21):6665−6673.
[60]Boeke JD,LaCroute F,Fink GR.A positive selection for mutants lacking orotidine-5'-phosphate decarboxylase activity in yeast:5-fluoro-orotic acid resistance.Mol Gen Genet,1984,197(2):345−346.
[61]Mizoguchi H,Tanaka-Masuda K,Mori H.A simple method for multiple modification of theEscherichiacoliK-12 chromosome. Biosci Biotechnol Biochem,2007,71(12):2905−2911.
[62]Tan Y,Xu D,Li Y,et al.Construction of a novel sacB-based system for marker-free gene deletion inCorynebacteriumglutamicum. Plasmid, 2012,67(1):44−52.
[63]Idiris A,Tohda H,Bi KW,et al.Enhanced productivity of protease-sensitive heterologous proteins by disruption of multiple protease genes in the fission yeastSchizosaccharomyces pombe.Appl Microbiol Biotechnol,2006,73(2):404−420.
[64]Posfai G,Kolisnychenko V,Bereczki Z,et al.Markerless gene replacement inEscherichia colistimulated by a double-strand break in the chromosome.Nucleic Acids Res,1999,27(22):4409−4415.
[65]Kolisnychenko V,Plunkett G,Herring CD,et al.Engineering a reducedEscherichia coligenome.Genome Res,2002,12(4):640−647.
[66]Posfai G,Plunkett G,Feher T,et al.Emergent properties of reduced-genomeEscherichia coli.Science,2006,312(5776):1044−1046.
[67]Sung BH,Lee CH,Yu BJ,et al.Development of a biofilm production-deficientEscherichia colistrain as a host for biotechnological applications.Appl Environ Microbiol,2006,72(5):3336−3342.
[68]Yu BJ,Kang KH,Lee JH,et al.Rapid and efficient construction of markerless deletions in theEscherichia coligenome.Nucleic Acids Res,2008,36(14):e84.
[69]Martinez-Garcia E,de Lorenzo V.Engineering multiple genomic deletions in Gram-negative bacteria:analysis of the multi-resistant antibiotic profile ofPseudomonas putidaKT2440.Environ Microbiol,2011,13(10):2702−2716.
[70]Suzuki N,Nonaka H,Tsuge Y,et al.Multiple large segmentdeletion method forCorynebacterium glutamicum.ApplMicrobiolBiotechnol,2005,69(2):151−161.
[71]Sternberg N, Sauer B, Hoess R, et al.Bacteriophage P1cregene and its regulatory region.Evidence for multiple promoters and for regulation by DNA methylation.J Mol Biol,1986,187(2):197−212.
[72]Leprince A,JanusD,de Lorenzo V,etal.Streamlining of aPseudomonas putidagenome using a combinatorial deletion method based on minitransposon insertion and the Flp-FRT recombination system.Methods Mol Biol,2012,813:249−266.
[73]Han Y,Li Y,Chen J,et al.Construction of monophosphoryl lipid a producingEscherichia colimutants and comparison of immuno-stimulatory activities of their lipopolysaccharides.Mar Drugs,2013,11(2):363−376.
[74]Fukiya S,Mizoguchi H,Mori H.An improved method for deleting large regions ofEscherichia coliK-12 chromosome using a combination of Cre/loxPand lambda Red.FEMS Microbiol Lett,2004,234(12):325−331.
[75]SuzukiN,Nonaka H,TsugeY,etal.New multiple-deletion method for theCorynebacterium glutamicumgenome,using a mutantloxsequence.Appl Environ Microbiol,2005,71(12):8472−8480.
[76]Goryshin IY,Naumann TA,Apodaca J,et al.Chromosomal deletion formation system based on Tn5 double transposition:use for making minimal genomes and essential gene analysis.Genome Res,2003,13(4):644−653.
[77]Yu BJ,Sung BH,Koob MD,et al.Minimization of theEscherichia coligenome using a Tn5-targeted Cre/loxPexcision system.Nat Biotechnol,2002,20(10):1018−1023.
[78]Mizoguchi H,Mori H,Fujio T.Escherichia coliminimum genome factory. Biotechnol Appl Biochem,2007,46(Pt3):157−167.
[79]Westers H,Dorenbos R,van Dijl JM,et al.Genome engineering reveals large dispensable regions inBacillus subtilis.Mol Biol Evol,2003,20(12):2076−2090.
[80]Leprince A,de Lorenzo V,Voller P,et al.Random and cyclical deletion of large DNA segments in the genome ofPseudomonasputida. Environ Microbiol,2012,14(6):1444−1453.
[81]Ara K,Ozaki K,Nakamura K,et al.Bacillusminimum genome factory:effective utilization of microbial genome information.Biotechnol Appl Biochem,2007,46(3):169−178.
[82]Lee JH,Sung BH,Kim MS,et al.Metabolic engineering of a reduced-genome strain ofEscherichiacolifor L-threonine production.Microb Cell Fact,2009,8(2):2.
[83]Csorgo B, Feher T, Timar E, et al.Low-mutation-rate,reduced-genomeEscherichia coli:an improved host for faithful maintenance of engineered genetic constructs.Microb Cell Fact,2012,11(1):1−13.
[84]Fukiya S,Mizoguchi H,Tobe T,et al.Extensive genomic diversity in pathogenicEscherichia coliandShigellastrainsrevealed by comparative genomic hybridization microarray.JBacteriol,2004,186(12):3911−3921.
[85]Sakihama Y,Mizoguchi H,Oshima T,et al.YdfH identified as a repressor ofrspAby the Use of reduced genomeEscherichia coliMGF-01.Biosci Biotechnol Biochem,2012,76(9):1688−1693.
[86]GoffM,Nikodinovic-Runic J,O'ConnorKE.Characterization of temperature-sensitive and lipopolysaccharide overproducing transposon mutants ofPseudomonas putidaCA-3 affected in PHA accumulation.FEMS Microbiol Lett,2009,292(2):297−305.
[87]Kim HJ,Lee CR,Kim M,et al.Dephosphorylated NPr of the nitrogen PTS regulates lipid A biosynthesis by directinteraction with LpxD.Biochem Biophys Res Commun,2011,409(3):556−561.
[88]Velazquez F,Pfluger K,Cases I,et al.The phosphotransferase system formed by PtsP,PtsO,and PtsN proteins controls production of polyhydroxyalkanoates inPseudomonas putida.J Bacteriol,2007,189(12):4529−4533.
[89]Wang XY, Quinn PJ. Lipopolysaccharide:Biosynthetic pathway and structure modification.Prog Lipid Res,2010,49(2):97−107.
[90]Mosberg JA,Gregg CJ,Lajoie MJ,et al.Improving lambda red genome engineering inEscherichia colivia rational removal of endogenous nucleases.PLoS One,2012,7(9):e44638.
[91]Jin DJ,Cagliero C,Zhou YN.Growth rate regulation inEscherichia coli.FEMS Microbiol Rev,2012,36(2):269−287.
[92]Yamaguchi Y,Inouye M.Regulation of growth and death inEscherichia coliby toxin-antitoxin systems. Nat Rev Microbiol, 2011, 9(11):779−790.
[93]Wang X,Lesterlin C,Reyes-Lamothe R,et al.Replication and segregation of anEscherichia colichromosome with two replication origins.Proc Natl Acad Sci USA,2011,108(26):E243−50.
[94]Wang RY,Shi ZY,Chen JC,et al.Cloning large gene clusters fromE.coliusinginvitrosingle-strand overlapping annealing.ACS Synth Biol,2012,1(7):291−295.
[95]Kuhlman TE,Cox EC.Site-specific chromosomal integration of large synthetic constructs.Nucleic Acids Res,2010,38(6):e92.
猜你喜欢
华人时刊(2023年1期)2023-03-14
林业科学(2022年1期)2022-03-23
汉字汉语研究(2021年2期)2021-08-30
计算机技术与发展(2021年6期)2021-07-06
中国蜂业(2021年5期)2021-05-22
汉字汉语研究(2019年2期)2019-08-27
特别健康(2018年2期)2018-06-29
电子制作(2017年17期)2017-12-18
浙江农林大学学报(2016年6期)2016-12-12
河北书画研究(2016年3期)2016-04-28
