
链霉菌无痕敲除方法研究进展
2013-09-04 08:35谷燕燕耿伟涛宋存江
生物工程学报 2013年8期
谷燕燕,耿伟涛,宋存江
南开大学生命科学学院 分子微生物与技术教育部重点实验室,天津 300071
链霉菌Streptomyces是革兰氏阳性菌 (G+),属于放线菌目 (Actinomycetes)链霉菌属,由于其能产生丰富的次级代谢产物,因此在工业生产中有着重要的作用。链霉菌在工业生产中主要用于抗生素的生产,目前许多天然来源的临床使用抗生素都是由链霉菌生产,如链霉素[1]、四环素、新霉素和氯霉素等[2]。另外链霉菌还用于生产其他产物如免疫抑制剂、抗肿瘤剂、抗虫剂及胞外水解酶,如蛋白酶、淀粉酶、几丁质酶、果胶酶、纤维素酶和木聚糖酶等[3]。由于其在工业上的重要作用,链霉菌遗传、发育及代谢调控的研究一直备受重视。
随着科技的发展,发酵工业与生物催化向人们提出了更高的要求,进一步提高目的产物的产量、纯度及开发有价值新化合物等成为工业生产急需解决的问题[4]。如何更快、更有效地对菌种进行改造,获得高产、高纯产物菌株成为我们研究的主要目标。应用传统的育种手段不但效率低、工作量大,而且获得的菌株遗传稳定性较差[5]。近年来随着分子生物学和基因组测序技术的不断发展以及合成生物学概念的提出[6],使人们能够利用已有的基因组信息和分子生物学方法目标明确地对目标菌株进行改造。我们一方面可以利用基因敲除技术阻断其他细胞代谢旁路来提高目的产物的产量和质量;另一方面还可以通过基因敲除或者基因修饰获得新的代谢产物从而达到微生物定向育种的目的[7]。此方法具有靶向性、安全性和稳定性的特点,逐渐成为发酵工业育种的重要手段。
1 链霉菌基因组基本特点
在众多的链霉菌菌株中,天蓝色链霉菌Streptomyce coelicolor是第一个完成全基因组测序的链霉菌,也是目前研究链霉菌遗传分化和抗生素生产的最好模式菌株[8]。目前已完成测序的链霉菌还有灰色链霉菌Streptomyce griseus[9]、阿维链霉菌Streptomyce avermitilis[10]等。链霉菌的染色体基因组呈线状,具有较高的GC含量,结构复杂,是目前已知的基因组最大的原核生物。其染色体中有一大小约6.28~6.5 Mb的核心区域,包含了链霉菌生长所必需的基因;线性染色体的两端均有长度大约1 Mb的亚端粒区,该区包含特殊的应变基因和次生代谢途径相关基因,如图1中所示S.griseus染色体示意图。同时链霉菌染色体的末端存在着大量反向重复序列,其在链霉菌发育过程中会缺失、扩增甚至环化,从而造成链霉菌染色体末端的不稳定[11]。
根据已有的链霉菌基因组测序结果分析显示,链霉菌体内编码天然产物生物合成途径的基因簇比己知的天然产物的基因簇多,这意味着链霉菌中许多的次生代谢产物未被发现,将其称为“隐蔽的”(Cryptic)基因簇,如图1(II)中的黑色条带,这是由于在实验室条件下只有少数的生物合成途径基因表达[12]。因此,可以通过有效的分子操作方法来对链霉菌基因组进行精简或对“隐蔽的”基因簇进行功能分析,以发挥链霉菌在生产发展中的巨大潜力。文中综述了近年来在链霉菌中应用的分子操作方法并重点对链霉菌的无痕敲除方法进行总结分析。
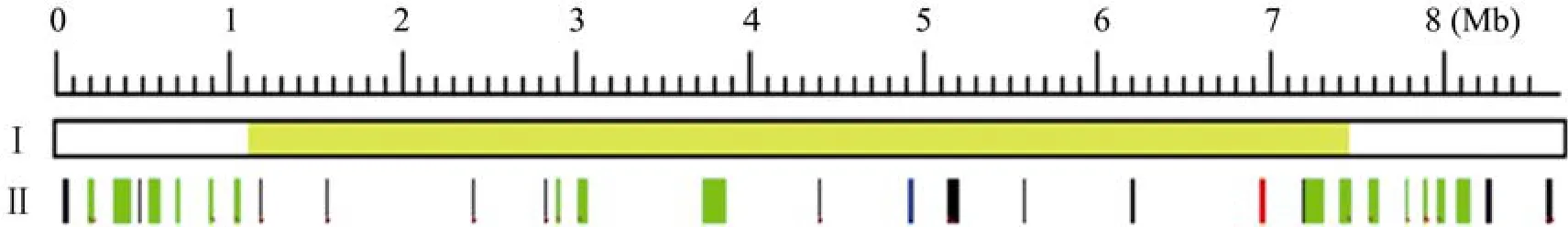
图1 S.griseus染色体示意图[9]Fig.1 Schematic representation of the S.griseus chromosome[9].(I)The yellow color indicates the “core region”.(II)Distribution of secondary metabolite gene clusters.Red:streptomycin(str,sts);blue:grixazone(gri);green:PKS and/or NRPS;black:other[9].
2 链霉菌有痕敲除方法
研究基因中断/敲除突变株的表型变化是一种广泛使用的研究功能未知基因的方法。链霉菌的有痕敲除方法是最早研究的一种基因敲除方法。由于在链霉菌中依赖于同源重组的基因敲除效率极低,当上下游同源臂小于1 kb时,几乎筛选不到双交换的目的菌株[13]。Gust等[14]将λ-red重组系统应用于构建链霉菌敲除载体,其构建过程如图 2。在含有λ-red重组系统的大肠杆菌Escherichia coliBW25113中,λ-red重组系统促进侧翼含有同源臂的抗性表达盒PCR扩增片段和含有链霉菌基因的粘粒发生同源重组,将得到的重组质粒通过接合转移的方法导入S.coelicolor中。由于其敲除载体的两端含有大片段的同源臂,重组粘粒和染色体之间可发生更高效的同源重组。但得到的目的菌株染色体上含有抗性筛选标记,不利于进行连续敲除。
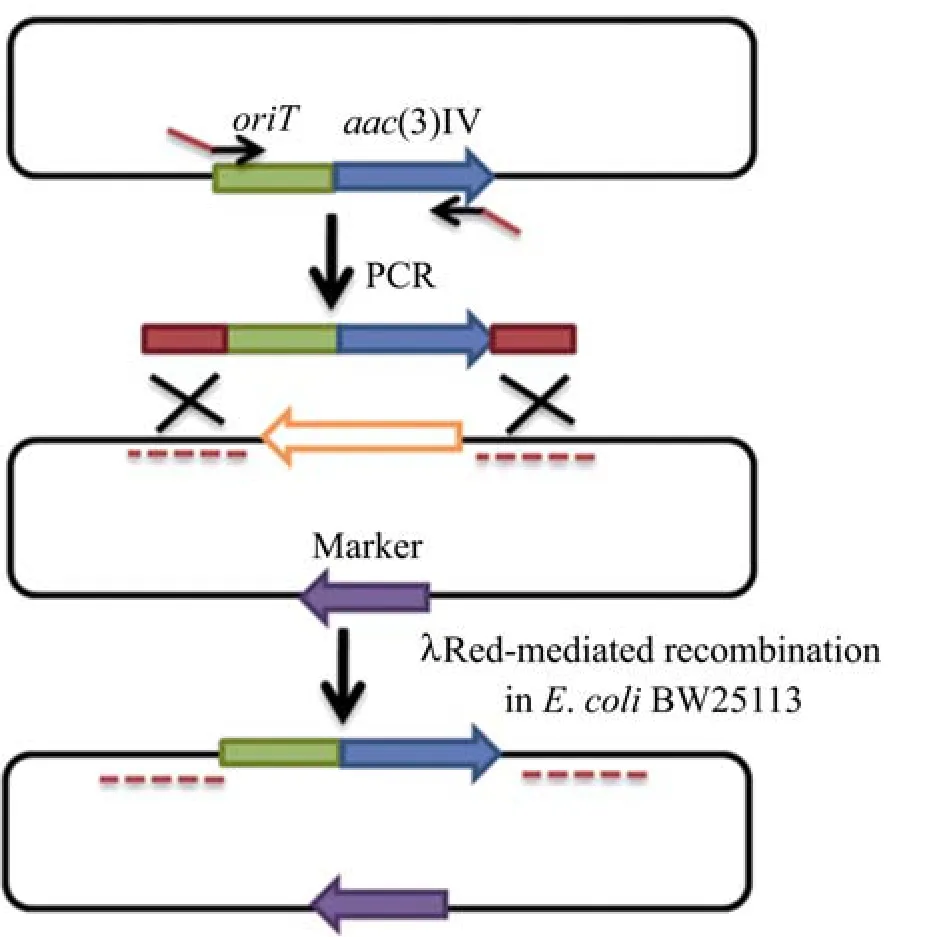
图2 应用λ-red重组系统构建敲除载体[14]Fig.2 Construction of knockout vector based on λ-red recombination system[14].
3 链霉菌的无痕敲除方法
链霉菌有痕敲除方法的缺点是每次敲除基因之后都会在染色体基因组上引入一个筛选标记,因此进行连续敲除时多种抗性标记在同一株菌内的叠加使用使得其后抗性标记的选择越发困难,而且抗性基因的插入还会影响其上下游基因的表达[15]。因此如何通过无痕敲除的方法对链霉菌进行基因操作成为人们研究的重点。
3.1 位点特异性重组
在后基因组时代,位点特异性重组系统已经成为高通量遗传分析中有利和高效的手段,在许多细菌和高等生物中已得到了广泛的应用[16]。位点特异性重组技术是通过对DNA的特定序列进行准确切割和重新连接,从而在基因或染色体水平上对生物进行改造的一种技术。利用位点特异性重组技术能够进行基因的定点插入[17]、外源基因的无痕表达[18]、基因的瞬态或定时的表达[19]和大片段敲除菌株的构建[20]等操作。目前在链霉菌中已得到应用的位点特异性重组系统有Cre/loxp系统、Dre/rox系统、Flp/FRT系统和Int-Xis系统 (表1)。下面将详细介绍应用不同的位点特异性重组系统构建无痕敲除突变株的方法。
3.1.1 Cre/loxp系统
Cre重组酶是1981年在P1噬菌体Phage P1中发现的一种重组酶,属于Int家族。Cre重组酶基因序列全长1 029 bp,蛋白大小为38 kDa。它能介导2个loxp位点之间的特异性重组,将2个loxp位点间的基因删除或倒位。loxp位点是Cre重组酶的特异性识别位点,是一段回文序列,由34 bp组成,即:2个13 bp的反向重复序列和1个8 bp的不对称间隔区[21]。
2006年,Khodakaramian等[25]将Cre重组酶在S.coelicolorA3(2)中成功表达,抗性基因的删除率达60%~90%。但是,由于Cre重组酶是通过ФC31噬菌体的转染进入链霉菌中,因此该方法未得到广泛的应用。Fedoryshyn等[26]在2008年对cre基因的密码子进行优化,合成了GC含量达67.7%的cre(a)基因,并使其在变铅青链霉菌Streptomyc lividans中以质粒形式成功地进行了表达,发现抗性基因的删除率可达100%。之前抗性基因敲除的研究多是应用该方法进行。
近年来,Cre/loxp位点特异性重组系统被逐渐应用于链霉菌的大片段无痕敲除。Komatsu等[27]应用Cre/loxp系统对S.avermitilis的基因组进行了精简,并且得到了一次敲除缺失 1.4 Mb的敲除突变株。在该方法中,2个loxp位点分别通过同源重组的方法整合到敲除基因的上下游,含有Cre重组酶基因的表达质粒pKU471通过接合转移的方法导入S.avermitilis中,在Cre重组酶的作用下,在2个loxp位点之间发生同源重组,最终完成靶序列的敲除,其敲除过程见图3。
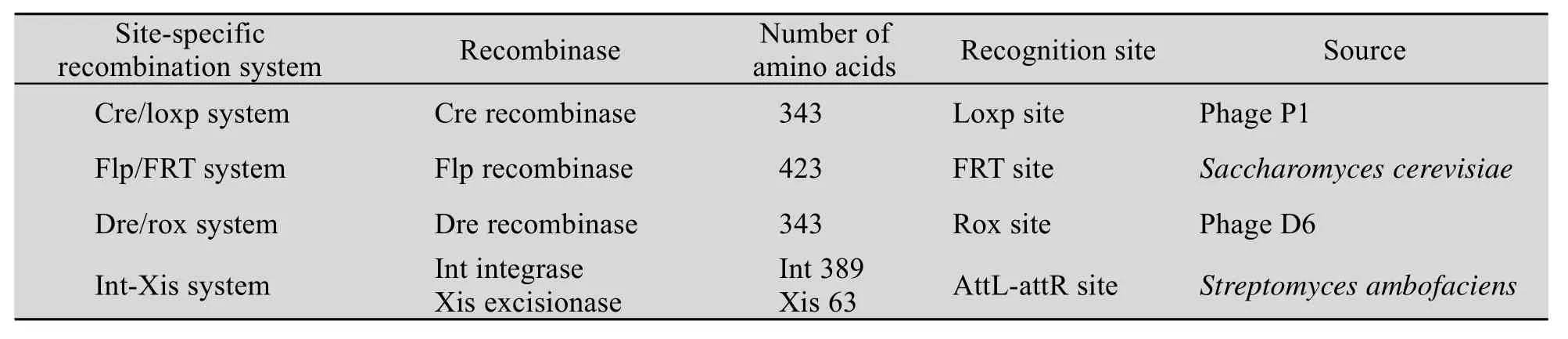
表1 链霉菌中应用的位点特异性重组系统分类[21-24]Table 1 Classification of Site-specific recombination system in Streptomyces[21-24]
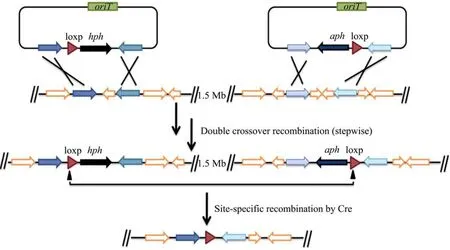
图3 Cre/loxp位点特异性重组系统构建无痕敲除突变株原理示意图[27]Fig.3 Strategy for construction of unmarked mutants based on Cre/loxp specific recombination system[27].
Herrmann等[28]对其敲除方法进行了改进,其敲除过程见图4。作者首先将2个loxp位点通过单交换的方法依次整合到敲除基因的上下游,在Cre重组酶的作用下将2个loxp位点间的靶基因删除,其敲除率可达100%。此方法和上述方法相比,其优点在于:此方法是通过单交换将loxp位点插入到靶基因的上下游,相较于双交换而言,单交换更容易获得。作者应用此方法分别将金色链霉菌Streptomyce aureofaciensTü117、链霉菌Streptomycessp.Tü6071和肉桂地链霉菌StreptomycecinnamonensisA519中的phenalinolactone(38 kb)、α-脂酶素 (67 kb)和莫能菌素 (83 kb)的合成酶基因簇进行了敲除,3株敲除突变株均不能产生相应的代谢产物。
应用Cre/loxp位点特异性重组系统的缺点是每进行一次敲除之后染色体上会留下1个loxp位点,连续进行多次敲除后会造成染色体的不稳定。为了维持染色体的稳定性,含有不同突变的lox位点 (loxLE和loxRE)的Cre/loxp系统已被广泛应用于真核微生物和细菌的敲除研究中[29]。理论上loxLE和loxRE重组后形成双突变的loxLERE位点,利用Cre重组酶不能很好地识别双突变的loxLERE位点的特性,因此可以在同一株菌中进行多次敲除。但是Herrmann等[28]发现在链霉菌中Cre重组酶对双突变的loxLERE位点仍有较好的识别能力,因此在链霉菌中即使使用Cre/loxLE-loxRE系统,仍然存在很高的染色体重组风险。
3.1.2 Flp/FRT系统
与Cre-loxP系统相似,Flp-FRT系统也是由一个重组酶和一段特殊的DNA序列组成。Flp重组酶基因全长1 272 bp,编码1个由423个氨基酸残基组成的多肽单体蛋白,该系统的另1个元件Flp识别位点 (Flp recognition target,FRT)与loxP位点也非常相似,同样由2个长度为13 bp的反向重复序列和1个长度为8 bp的核心序列构成。在该系统发挥作用时,2个FRT位点的方向性决定了目的片段的缺失或是倒转。与Cre相似,Flp发挥作用也不需要任何辅助因子,同时在不同的条件下具有良好的稳定性[22]。
由于flp基因GC含量太低,无法在链霉菌中表达来进行位点特异性重组,Gust等[14]巧妙地将Flp-FRT位点特异性重组系统和λ-red重组系统结合在一起来对链霉菌的基因进行敲除。作者将该方法命名为PCR-Targeting(图5)。
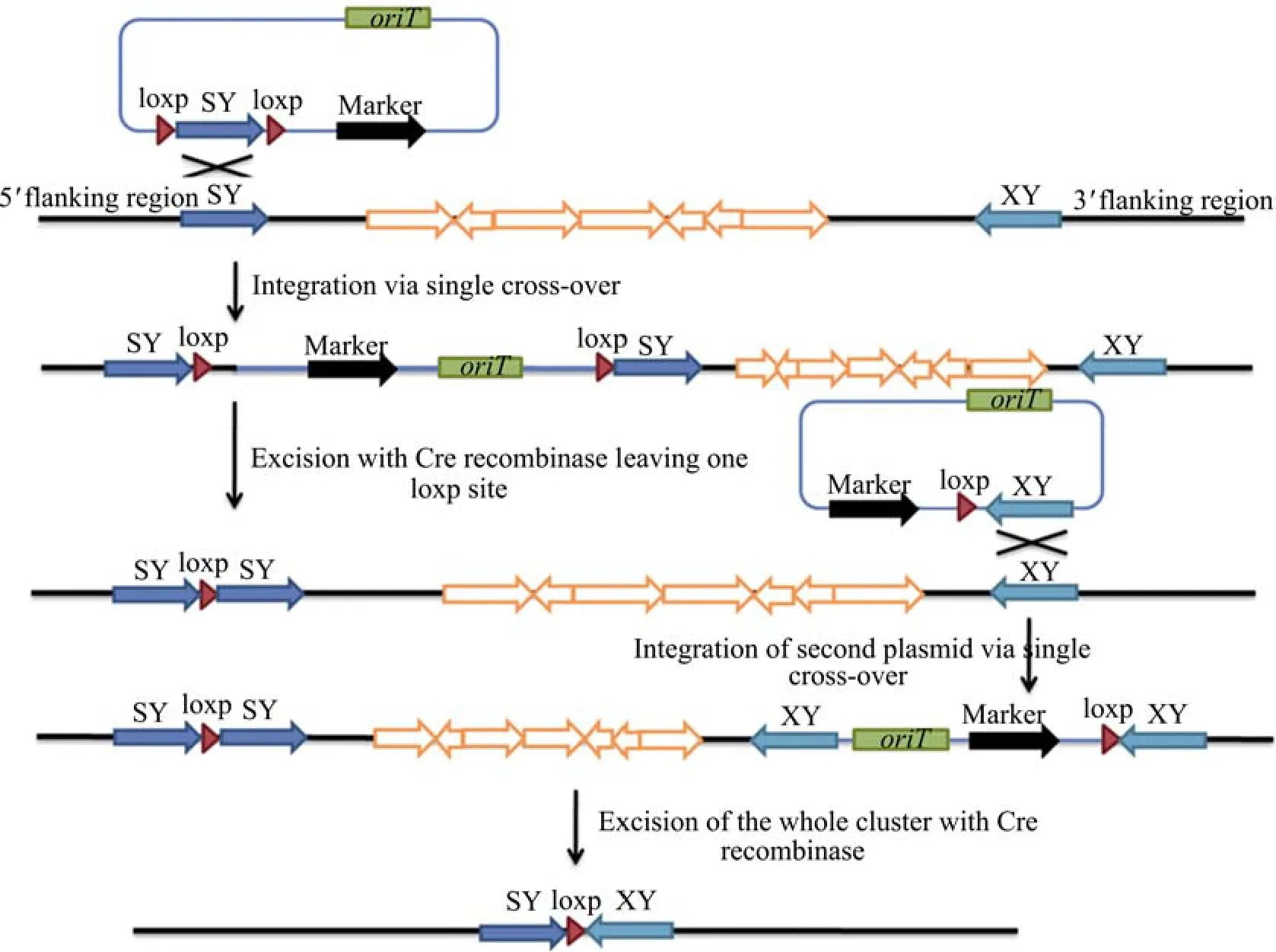
图4 改进后的Cre/loxp位点特异性重组系统构建无痕敲除突变株原理示意图[28]Fig.4 Strategy for construction of unmarked mutants based on improved Cre/loxp specific recombination system[28].
文章中作者应用PCR-Targeting方法来分析在S.coelicolorA3(2)中土味素生物合成所必需的基因。首先通过序列比对发现S.coelicolorA3(2)含有两个倍半萜烯合酶基因——cyc1和cyc2,然后用该方法将二者分别框内敲除后发现当敲除cyc2时,将不产生土味素。对cyc2编码的蛋白分析发现:Cyc2含有两个相似的倍半萜烯合酶结构域。为了验证该两个结构域是否都是土味素生物合成所必需的结构域,作者分别对Cyc2的C端和N端进行了框内敲除。结果表明只有Cyc2的N端是土味素合成所必需的基因。成功地应用PCR-Targeting方法将S.coelicolor基因组中大小为4~7 kb的100多个片段进行敲除。
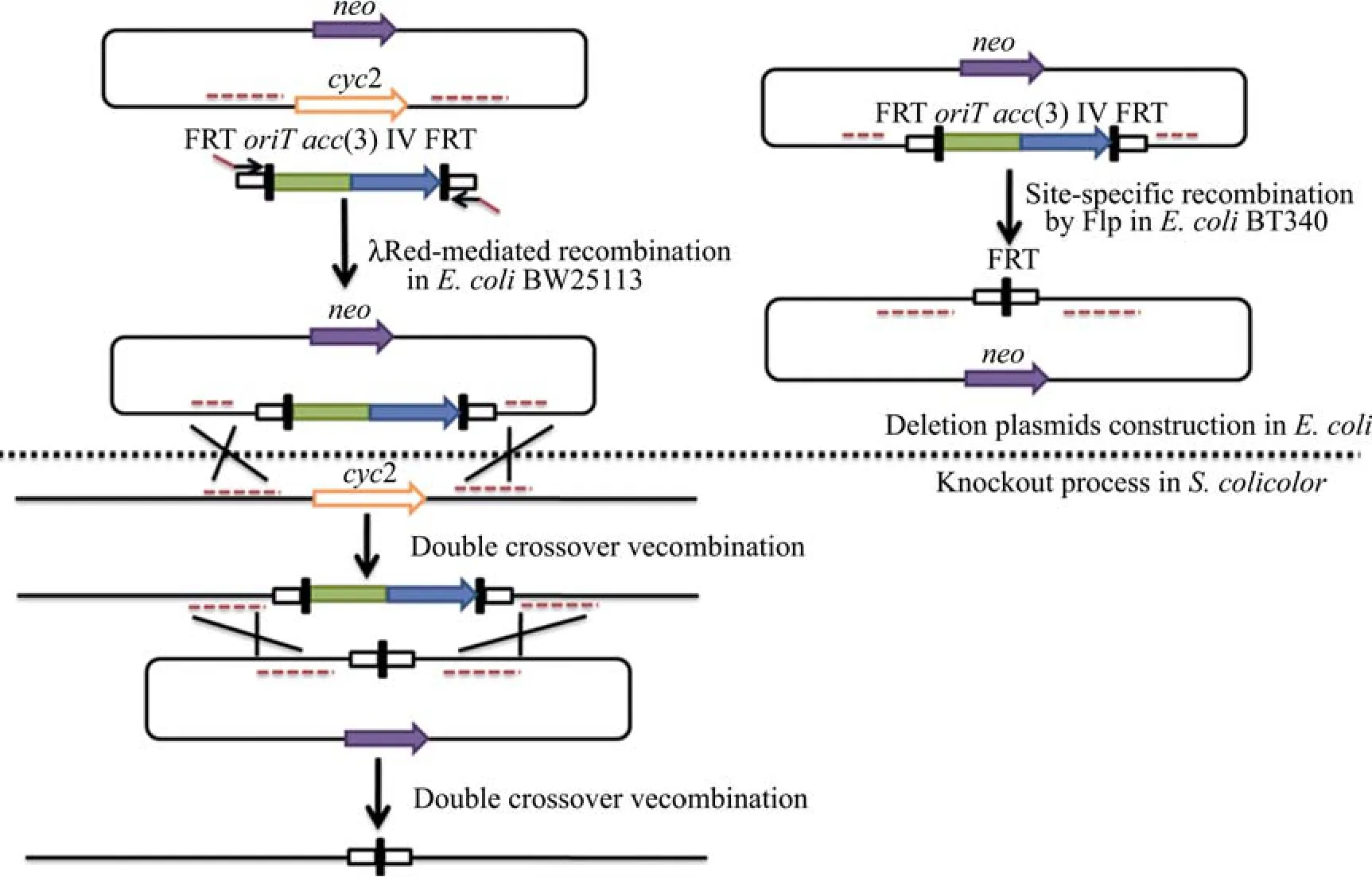
图5 PCR-Targeting构建无痕敲除突变株原理示意图[14]Fig.5 Strategy for construction of unmarked mutants utilizing PCR-Targeting[14].
2008年,Fedoryshyn等[30]合成 GC含量达60.6%的flp基因,其能在S.coelicolorM145、S.lividansTK24和西班牙糖丝菌Saccharothrix espanaensis中表达。然而,其将染色体上两边含有FRT位点的阿伯拉霉素抗性基因的删除率仅达40%,效率要远低于Cre/loxp系统的效率。该方法虽然效率相对较低,但还是可以相对容易地获得无痕敲除突变株。
3.1.3 Dre/rox系统
2004年,Sauer和 McDermott[23]对 4株 P1相关噬菌体 (P7噬菌体Phage P7、p15B缺陷噬菌体 Defective phage p15B、ψw39转导噬菌体Transducing phage ψw39和 D6转导噬菌体Transducing phage D6)的7 kb immc区进行比对发现了1个新的DNA重组酶——Dre。D6位点特异性重组酶 (Dre)和Cre重组酶相似均是酪氨酸重组酶,但其识别的是32 bp的DNA序列rox。Dre重组酶和Cre重组酶具有异种特异性,Dre重组酶不能识别loxp位点,同时Cre重组酶也不能识别rox位点。
为使Dre重组酶在链霉菌中进行表达,Herrmann等[28]将其编码基因的 GC含量进行优化,基因合成后分别克隆到pAL1和pUWLoriT构建成pALDRE和pUWLDRE。为研究Dre/rox系统的重组效率,作者分别在小单胞菌属Micromonosporasp.Tü6368 、Saccharothrix espanaensis、S.coelicolorT3456-20和S.lividansTK24中进行了抗性基因删除试验,结果发现在不同菌株中抗性基因删除率均可达100%,说明Dre成功表达并且 Dre/rox系统有很高的重组效率,这就为放线菌的基因操作提供了新的工具——Dre/rox位点特异性重组系统。
3.1.4 Int/Xis系统
Raynal等[24]利用产二素链霉菌Streptomyces ambofaciens中原始质粒pSAM2构建了一种新型的链霉菌敲除体系——Int/Xis位点特异性重组系统。pSAM2为链霉菌整合质粒,具有和温和噬菌体位点特异性重组相似的系统,其编码的复制酶、切除酶和整合酶基因repSA、xis和int是以操纵子的形式存在。整合酶促进attP位点和attB位点间的重组,从而将外源片段整合到染色体上并且形成attL和attR位点。Int/Xis系统的基本原理是:当整合酶和删除酶同时表达时可以促进attL位点和attR位点之间发生分子内位点特异性重组,从而将两位点间的序列删除,达到敲除的目的。
作者首先构建了一个attL-抗性基因-attR的基因盒和一系列含int和xis基因的表达载体(pSOV507和pSOV508)。两端带有EcoRV平末端内切酶的attL-抗性基因-attR基因盒,可以平末端的形式插入到任何克隆基因中,其敲除载体及敲除过程见图6。S.ambofaciens中螺旋霉素合成酶基因簇中含有7个ORF,编码的酶参与了螺旋霉素生物合成的不同步骤。作者应用该系统将S.ambofaciens中螺旋霉素合成酶基因簇中的ORF3进行了框内敲除。其具体操作过程如下:首先构建了敲除载体pOSV513——用attL-hyg-attR的基因盒置换了ORF3中的270 bp核苷酸序列。然后将pOSV513导入S.ambofaciens,通过筛选双交换得到S.ambofaciensorf3::att1_hyg菌株。最后将表达载体pSOV508导入上述菌株,表达Int和Xis促进attL位点和attR位点之间发生特异性重组,将两位点间的抗性基因删除,在染色体上留下att1位点。
3.2 大范围核酸内切酶
同源重组是一种广泛存在于微生物中的生物事件,涉及的分子事件是高度保守,包含了DNA的双链断裂和修复。以同源重组为基础的分子操作方法是基因组工程的强大工具,它已广泛应用于不同生物中基因的敲除、替换或外源基因的插入。在天蓝色链霉菌中已建立了各种遗传工具,如常规的依赖于同源重组的基因破坏和基因替换以及更有效的PCR-targeting方法。然而,即使应用PCR-targeting方法在天蓝色链霉菌中获得无痕突变仍然是个费时的过程。
Rouet和Choulika等的研究发现在特定位点的双链DNA断裂可促进同源重组发生的概率[31-32]。大范围核酸内切酶可造成 DNA的双链断裂,它能够特异性识别>12 bp的核苷酸序列。归巢核酸内切酶是大范围核酸内切酶的代表,参与内含子归巢的过程,广泛存在于低等真核生物、细菌和古菌中[33]。类似于Ⅱ型限制性内切酶,归巢核酸内切酶具有很高的序列特异性,但Ⅱ型限制性内切酶识别短核苷酸序列 (3~8 bp),而归巢内切酶识别较长核苷酸序列 (12~40 bp)[34]。
3.2.1Ⅰ-SceⅠ大范围核酸内切酶
大范围核酸内切酶Ⅰ-SceⅠ是在酿酒酵母Saccharomyces cerevisiae的线粒体中发现的,该核酸内切酶含有235个氨基酸,识别18 bp核苷酸序列 (5′-TAGGGATAACAGGGTAAT-3′)[35-36]。理论上700亿bp随机序列可能出现1个18 bp识别序列,目前所分析的链霉菌基因组序列中未发现Ⅰ-SceⅠ识别位点,因此避免了基因组被大范围核酸内切酶破坏的风险。Ⅰ-SceⅠ核酸内切酶已被应用为链霉菌染色体双链断裂的诱导剂,并且利用该系统证明了在链霉菌中同源重组是DNA双链断裂的主要修复机制[37]。应用以Ⅰ-SceⅠ核酸内切酶为基础的基因操作方法已成功在E.coli[38],炭疽芽胞杆菌Bacillus anthracis[39],类鼻疽伯克氏菌Burkholderia pseudomallei[40],谷氨酸棒状杆菌Corynebacterium glutamicum[41]和铜绿假单胞菌Pseudomonas aeruginosa[42]中构建了缺失突变株。
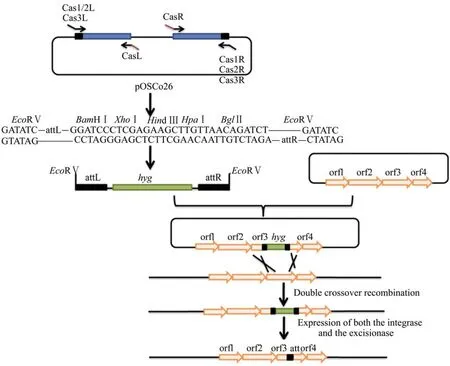
图6 Xis/Int位点特异性重组系统构建无痕敲除突变株原理示意图[24]Fig.6 Strategy for construction of unmarked mutants based on Xis/Int specific recombination system[24].
由于原始的Ⅰ-SceⅠ编码基因GC含量只有20%,无法在链霉菌中进行表达。Siegl等[37]和Lu等[43]将其GC含量进行优化,最终的Ⅰ-SceⅠ编码基因GC含量分别达56.6%和55.2%,合成后克隆到表达载体,形成表达载体pLU101、pALSCE和 pUWLSCE。Lu等应用优化后的Ⅰ-SceⅠ核酸内切酶基因敲除方法将S.coelicolor中的放线紫红素合成酶基因簇中约20 kb的片段进行了敲除。首先构建了含有靶基因上下游同源臂、抗性筛选标记和1个Ⅰ-SceⅠ识别位点的自杀敲除质粒,该质粒通过接合转移导入到链霉菌中,并通过单交换整合到染色体上。随后将含有Ⅰ-SceⅠ核酸内切酶基因的表达载体导入到上述突变株中,Ⅰ-SceⅠ核酸内切酶表达后识别18 bpⅠ-SceⅠ位点,并在该位点将双链DNA切断促使双交换的发生。最后通过PCR检测可很容易筛选到敲除突变株 (图7)。该方法的优点是效率高且Ⅰ-SceⅠ核酸内切酶不仅能够促进同源重组而且是嵌合体的反向筛选标记,同时得到的敲除突变株是无痕的,可以在同一株菌中连续进行多次敲除实验。
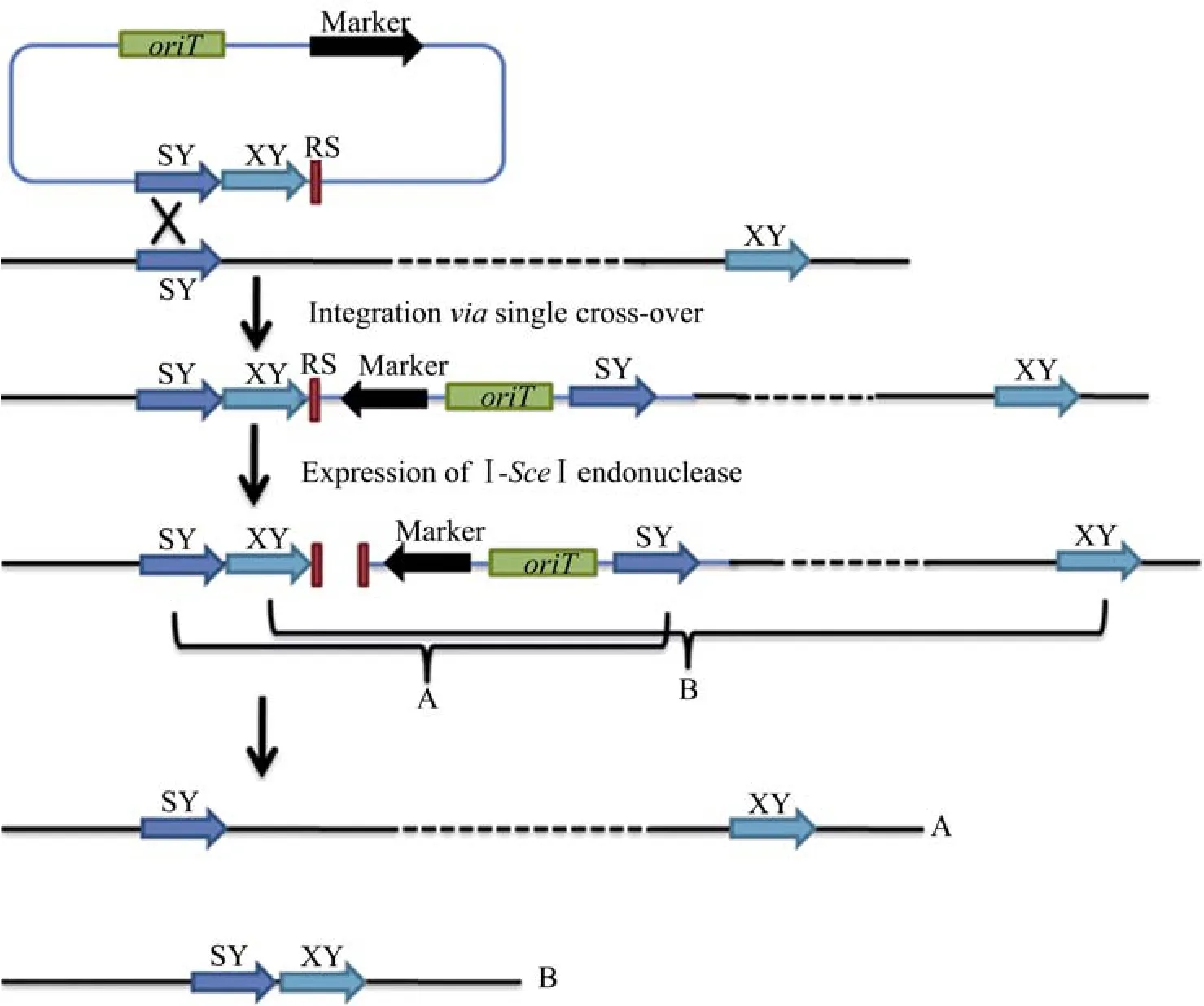
图7Ⅰ-SceⅠ核酸内切酶构建无痕敲除突变株原理示意图[37,43]Fig.7 Strategy for construction of unmarked mutants based on I-SceI endonuclease[37,43].
4 展望
链霉菌能产生大量的具有重要价值的天然代谢产物,因此链霉菌的遗传发育与次级代谢领域的任何进展都与人类生活息息相关。笔者所在研究室分离到1株可产ε-聚赖氨酸的小白色链霉菌Streptomyces albulusNK660[44],正在进行该菌株的全基因组测序。通过采用pUC19和pAL1质粒,并结合Cre/loxp位点特异性重组的特性,构建了适合S.albulusNK660菌株大片段无痕敲除的Cre/loxp方法。同时采用pUC19和pKC1139质粒,结合Ⅰ-SceⅠ大范围核酸内切酶的特点,构建了Ⅰ-SceⅠ的大片段无痕敲除方法。Cre/loxp和Ⅰ-SceⅠ的大片段无痕敲除原理示意图见图4和图7。笔者期望通过构建优势小基因组菌株来提高目的产物的产量,减少其他次级代谢产物的含量并降低原料底物消耗量。相信随着大片段无痕敲除链霉菌冗余基因方法的不断完善,必将推进链霉菌的基础研究和相关次级代谢产物工业的发展。
[1]Schatz A,Bugie E,Waksman SA.Streptomycin,a substance exhibiting antibiotic activity against gram-positive and gram-negative bacteria.Proc Soc Exp Biol Med,1944,55:66−69.
[2]Wang S, Gao YL. Research progress of aminoglycoside drug.Pharm J Chin PLA,2009,25(4):329−333(in Chinese).
王舒,高永良.氨基糖苷类药物研究新进展.解放军药学学报,2009,25(4):329−333.
[3]Li O,Miao KP.Progress inStreptomycessecondary metabolism.Chin J Antibiotics,2005,30(11):695−698(in Chinese).
李欧,缪克排.链霉菌次级代谢研究进展.中国抗生素杂志,2005,30(11):695−698.
[4]Zhang HY,Shen NK,Zhou X.Gene knockout technology and its application in microbial breeding. Liquor-Making Sci Technol, 2010,190(4):21−25(in Chinese).
张红岩,申乃坤,周兴.基因敲除技术及其在微生物育种中的应用.酿酒科技,2010,190(4):21−25.
[5]Parekh S,Vinci VA,Strobel RJ.Improvement of microbial strains and fermentation processes.Appl Microbiol Biotechnol,2000,54(3):287−301.
[6]Heinemann M,Panke S.Synthetic biology-putting engineering into biology.Bioinformatics,2006,22(22):2790−2799.
[7]Song AD,Zhang SS,Wang FQ,et al.Gene knockout and its applications in industrial microbe breeding.J Biol,2011,28(6):68−72(in Chinese).
宋安东,张沙沙,王风芹,等.基因敲除在工业微生物育种方面的应用.生物学杂志,2011,28(6):68−72.
[8]Bentley SD,Chater KF,Challis GL,et al.Complete genome sequence ofthe modelactinomyceteStreptomycescoelicolorA3(2).Nature,2002,417(6885):1141−1147.
[9]Ohnishi Y,Ishikawa J,Hara H,et al.Genome sequence of the streptomycin-producing microorganismStreptomyces griseusIFO 13350.J Bacteriol,2008,190(11):4050−4060.
[10]Ikeda H,Ishikawa J,Hanamoto A,et al.Complete genomesequencecomparativeanalysisofthe industrial microorganismStreptomyces avermitilis.Nat Biotechnol,2003,21(5):526−531.
[11]Wu XC,Miao KP,Qian KX.Advances of genome and secondary metabolism inStreptomyces.Acta Genet Sin,2005,32(11):1221−1227(in Chinese).
吴雪昌,缪克排,钱凯先.链霉菌基因组及次生代谢研究进展.遗传学报,2005,32(11):1221−1227.
[12]Zhou M,Jing XY,Xie PF,et al.Sequential deletion of all the polyketide synthase and nonribosomal peptide synthetase biosynthetic gene clusters and a 900-kb subtelomeric sequence of the linear chromosome ofStreptomyces coelicolor.FEMS Microbiol Lett,2012,333(2):169−179.
[13]Kieser T,Bibb MJ,Buttner MJ,et al.PracticalStreptomycesGenetics.United Kingdom:the John Innes Foundation Press,2000:311−315.
[14]Gust B,Challis GL,Fowler K,et al.PCR-targetedStreptomycesgene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin.Proc Natl Acad Sci USA,2003,100(4):1541−1546.
[15]Siegl T,Luzhetskyy A.Actinomycetes genome engineering approaches. Antonie Van Leeuwenhoek,2012,102(3):503−516.
[16]Branda CS,DymeckiSM.Talking abouta revolution:the impact of site-specific recombinases on genetic analyses in mice.Dev Cell,2004,6(1):7−28.
[17]Kuhstoss S,Richardson MA,Rao RN.Plasmid cloning vectors that integrate site-specifically inStreptomycesspp..Gene,1991,97(1):143−146.
[18]Schweizer HP.Applications of theSaccharomyces cerevisiaeFlp-FRT system in bacterial genetics.J Mol Microbiol Biotechnol,2003,5(2):67−77.
[19]Slauch JM,Camilli A.IVET and RIVET:use of gene fusions to identify bacterial virulence factors specifically induced in hosttissues.Methods Enzymol,2000,326:73−96.
[20]SuzukiN,Nonaka H,Tsuge Y,etal.New multiple-deletion method for theCorynebacterium glutamicumgenome,using a mutant lox sequence.Appl Environ Microbiol,2005,71(12):8472−8480.
[21]Sternberg N,Hamilton D.Bacteriophage P1 site-specific recombination. I. Recombination between LoxP sites.J Mol Biol,1981,150(4):467−468.
[22]Volkert FC, Wilson DW, Broach JR.Deoxyribonucleic acid plasmids in yeasts.Microbiol Rev,1989,53(3):299−317.
[23]Sauer B,McDermott J.DNA recombination with a heterospecific Cre homolog identified from comparison of the pac-c1 regions of P1-related phages. Nucleic Acids Res, 2004, 32(20):6086−6095.
[24]Raynal A,Karray F,Tuphile K,et al.Excisable cassettes:new tools for functional analysis ofStreptomycesgenomes.Appl Environ Microbiol,2006,72(7):4839−4844.
[25]Khodakaramian G,Lissenden S,Gust B,et al.Expression of Cre recombinase during transient phage infection permits efficient marker removal inStreptomyces.Nucleic Acids Res,2006,34(3):e20.
[26]Fedoryshyn M,Welle E,Bechthold A,et al.Functional expression of the Cre recombinase in actinomycetes.Appl Microbiol Biotechnol,2008,78(6):1065−1070.
[27]Komatsu M,Uchiyama T,Omura S,etal.Genome-minimizedStreptomyceshostfor the heterologous expression of secondary metabolism.Proc Natl Acad Sci USA, 2010, 107(6):2646−2651.
[28]Herrmann S,SieglT,Luzhetska M,etal.Site-Specific recombination strategies for engineering actinomycete genomes.Appl Environ Microbiol,2012,78(6):1804−1812.
[29]Leibig M,Krismer B,Kolb M,et al.Marker removal inStaphylococcivia Cre recombinase and different lox sites.Appl Environ Microbiol,2007,74(5):1316−1323.
[30]Fedoryshyn M,Petzke L,Welle E,et al.Marker removal from actinomycetes genome using Flp recombinase.Gene,2008,419(1/2):43−47.
[31]RouetP,Smih F,Jasin M.Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease.Mol Cell Biol,1994,14(12):8096−8106.
[32]Choulika A,Perrin A,Dujon B,et al.Induction of homologous recombination in mammalian chromosomes by using the I-SceI system ofSaccharomyces cerevisiae.Mol Cell Biol,1995,15(4):1968−1973.
[33]Jasin M.Genetic manipulation of genomes with rarecutting endonucleases.Trends Genet,1996,12(6):224−228.
[34]Chevalier BS, Stoddard BL. Homing endonucleases:structural and functional insight into the catalysts of intron/intein mobility.Nucleic Acids Res,2001,29(18):3757−3774.
[35]Monteilhet C, Perrin A,Thierry A, et al.Purification and characterization of thein vitroactivity of I-SceI,a novel and highly specific endonuclease encoded by a group I intron.Nucleic Acids Res,1990,18(6):1407−1413.
[36]Plessis A,Perrin A,Haber JE,et al.Site-specific recombination determined by I-SceI, a mitochondrial group I intron-encoded endonuclease expressed in the yeast nucleus.Genetics,1992,130(3):451−460.
[37]SieglT,Petzke L,Welle E,etal.I-SceI endonuclease:a new tool for DNA repair studies and genetic manipulations inStreptomycetes.Appl Microbiol Biotechnol,2010,87(4):1525−1532.
[38]Posfai G,Kolisnychenko V,Bereczki Z,et al.Markerless gene replacement inEscherichia colistimulated by a double-strand break in the chromosome.Nucleic Acids Res,1999,27(22):4409−4415.
[39]Janes BK,Stibitz S.Routine markerless gene replacement inBacillus anthracis.Infect Immun,2006,74(3):1949−1953.
[40]Lopez CM,Rholl DA,Trunck LA,et al.Versatile dual-technology system for markerless allele replacement inBurkholderia pseudomallei.Appl Environ Microbiol,2009,75(20):6496−6503.
[41]Suzuki N,Nonaka H,Tsuqe Y,et al.Multiple large segmentdeletion method forCorynebacteriumglutamicum.ApplMicrobiolBiotechnol,2005,69(2):151−161.
[42]Wong SM,Mekalanos JJ.Genetic footprinting with marinerbased transposition inPseudomonas aeruginosa.Proc Natl Acad Sci USA,2000,97(18):10191−10196.
[43]Lu ZQ,Xie PF,Qin ZJ.Promotion of markerless deletion of the actinorhodin biosynthetic gene cluster inStreptomyces coelicolor.Acta Biochim Biophys Sin,2010,42(10):717−721.
[44]Song CJ,Geng WT,Wang SF,et al.Streptomycessp. NK660 (Streptomycesalbulus) and its fermentation cultivation method of ε-poly-lysine:CN,201110362088.8.2011-10-03(in Chinese).
宋存江,耿伟涛,王淑芳,等.链霉菌 sp.NK660(Streptomyces albulus)及其用于ε-聚赖氨酸的发酵培养方法:CN,201110362088.8.2011-10-03.
猜你喜欢
当代水产(2022年1期)2022-04-26
今日农业(2021年11期)2021-08-13
中西医结合肝病杂志(2020年2期)2020-10-27
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
中成药(2018年7期)2018-08-04
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
少儿科学周刊·少年版(2015年3期)2015-07-07
少儿科学周刊·少年版(2015年3期)2015-07-07
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
