
双碳醇水溶液的1H NMR实验与理论分析
2017-11-01 18:11唐景春
物理化学学报 2017年10期
叶 斌 张 健 高 才 ,* 唐景春
(1合肥工业大学汽车与交通工程学院,合肥 230009;2合肥工业大学电气与自动化工程学院,合肥 230009)
双碳醇水溶液的1H NMR实验与理论分析
叶 斌1张 健2高 才1,*唐景春1
(1合肥工业大学汽车与交通工程学院,合肥 230009;2合肥工业大学电气与自动化工程学院,合肥 230009)
采用核磁共振氢谱(1H NMR)和量子化学(QC)方法研究不同温度下乙醇水溶液和乙二醇水溶液中醇与水之间的相互作用。观察实验结果发现两种醇水溶液中水质子的化学位移呈现两种不同的变化趋势。随着含水量的增加,乙醇(ET)水溶液中水质子化学位移急剧降低,而乙二醇(EG)水溶液中水质子的化学位移缓慢增加。两种醇水溶液中的羟基质子随着浓度的增加,其共振峰移向低场。不同温度下随着浓度的增加两种醇水溶液的烷基质子共振峰单调的移向低场。几何结构优化结果表明醇羟基质子与水质子之间氢键的形成弱化了醇中 O―H键,从而导致其键长增加。值得注意的是在相同的极化作用和扩散作用下采用密度泛函理论(DFT)(B3LYP)计算得到的ET和EG的C―H键,C―C键和O―H键的键长值大于采用HF理论计算得到的结果。与此相反的是采用HF理论得到的ET和EG的O―H···O键强度大于采用DFT(B3LYP)理论得到的结果。几何构型优化结果与实验结果相吻合。在NMR化学位移的计算中,就文中所提到的理论水平而言,DFT要优于HF。而对于同一理论,其基组越大,计算值越接近实验值。
乙醇;乙二醇;核磁共振氢谱;化学位移;Hartree-Fork理论;密度泛函理论
1 Introduction
Alcohol-water mixtures have been intensively studied and the researches on them are currently of wide scientific interest.Both ethanol (ET) and ethylene glycol (EG) are small molecule alcohols with two carbons. However, the different number of hydroxyl makes ET and EG show distinguishing properties.The chemical structures of ET (monohydric alcohol) and EG(dihydric alcohol) are shown in Fig.1. The unique structure of alcohols makes it possible to systematically study the solute and water interactions. To understand the structure and dynamics of alcohol-water mixtures, all sorts of theoretical and experimental methods have been carried out1−14. Gao et al.6,7has studied the cooperative relaxation in 1,2-propanediol and D-sorbitol. Sengwa et al.8has studied the formation of hydrogen bonds between different types of molecules in binary alcohol mixtures by an analysis of their dielectric parameters.Zhang et al.10,11has performed Molecule Dynamics (MD)Simulation to investigate the aqueous binary mixtures of alcohols. Ramam et al.14has probed the intermolecular association through hydrogen bonding in aqueous solutions of glycerol by ultrasonic and density functional theory (DFT).
Although ET and EG aqueous solutions have been experimentally and theoretically explored15−19, few literature focuses on comparing ET and EG aqueous solutions. ET and EG have the same number of carbon, but the interactions between solute and water show distinctly different in the two alcohol aqueous solutions. This may be due to the H-bonds formed between alcohol and water. Thus, a comparison of the hydrogen bonds formation ability between ET and EG is necessary to understand the solvent properties of ET-water and EG-water mixtures on the molecular scale. There are three kinds of chemically different protons and two kinds of chemical different protons in the ET molecule and EG molecule, respectively. Since the composition dependence of the chemical shift of the water proton and alcohol proton in the mixtures yields information on the polarization of solute and water molecules and the hydrogen-bonding strength, the results of1H Nuclear Magnetic Resonance (NMR) measurements well help to understand the alcohol-water mixtures. In this paper, the NMR technology will be used to probe the micro-structures of the alcohol and water interactions.
Recently, quantum chemical theoretical computation has played an important role in treating chemical problems and interpreting chemical phenomena20−26. By different theory levels, methods and basis sets, many problems related to chemistry have been solved. Because the NMR spectrum is influenced by both solvation and conformational effects, the geometrical optimization calculations and NMR chemical shift prediction help to unravel the different contributions and understand the H-bond in mixtures in many applications of quantum chemistry. However, Because of the difference of the theoretical level, the computation values of chemical shift are not exactly consistent with those obtained by experiment. In the prediction of NMR magnetic shielding constants, DFT and Hartree-Fork (HF) are the most commonly used theoretical levels. In order to improve the speed and accuracy of quantum chemical NMR theoretical calculation, by comparing the experimental value with the theoretical value of the NMR chemical shift obtained by using different basis sets at DFT and HF levels of theory, respectively, this paper explores the better method of calculating double alcohols NMR chemical shift.

Fig.1 Chemical structures of ET and EG molecules (a) chemical formula of ET (b) chemical formula of EG.
In this article, the1H NMR measurement and quantum chemistry theoretical method were used to study the small molecular double-carbon alcohols, which will be help to understand the microstructures of EG and ET and discriminate the macroscopic physical and chemical properties. Recently,small molecular alcohols and water mixtures are often used as cryoprotective agent (CPA). If a better understanding of the interactions involved between water and alcohols can be achieved, a better effect can be reached.
2 Experiment and theory
2.1 Materials and method
Ethanol (ET, 99%, J&K Chemical LTD.) and ethylene glycol(EG, 99%, J&K Chemical LTD.) were purified by distillation and solutions were prepared by mixing appropriate amounts of solute and distilled water. The samples were placed in the lab for 2 h for complete homogeneity. A Bruker-AV400 NMR spectrometer (Switzerland) was used for chemical shift measurements.
The samples were sucked into a 2-mm capillary (NORELL,America) and sealed at both ends. The capillary was placed in a 5-mm NMR tube (NORELL, America) containing CDCL3(locking liquid) and TMS (external reference). The temperature was controlled at 290.15, 300.15 and 310.15 K, respectively.
As using the external27TMS solution, the susceptibility corrections are necessary. These were made by using volume susceptibilities. The following relation was used28:

where x represents the mole fraction of solute and water; and χ is the diamagnetic susceptibility, which can be found in the compilations of Selwood30; δcorrepresents the correcting value;δexpis the experimental value.
2.2 Quantum chemical calculation
2.2.1 Theory
The theoretical prediction of magnetic shielding constants is based on perturbation theory. As a result of the perturbation analysis, the magnetic shielding and the magnetic susceptibilities can be obtained31−33. The magnetic shielding tensor for a given nucleus k is composed of the elements34:

where μ represents the magnetic moment of a nucleus. α and β are the components of the external magnetic field and induced magnetic moment, respectively. Because the calculated magnetic properties depend on the gauge origin for any incomplete basis set, the magnetic shielding constant is very difficult to compute31−33. Therefore, the choice of electronic structure calculation scheme and the algorithm used to predict gauge-invariant magnetic shielding constants must be made in the theoretic prediction of NMR chemical shift. In quantum chemistry computation, the methods associated with NMR include GIAO (gauge including atomic orbital), IGAIM(individual gauges for atoms in molecules), IGLO (individual gauge for localized orbitals) and CSGT (continuous set of gauge transformation) at each theory level. GIAO is considered to be the preferred method because of the faster converges of GIAO. In this paper, GIAO method is adopted.
The theoretical calculation of NMR usually uses Hartree-Fock (HF) level of theory and DFT/B3LYP level. Both HF and DFT are based on the ab initio principles which is the basic theory of quantum mechanics. The HF level of theory is based on the Hartree-Fork equation35:

The HF and DFT levels of theory have their own advantages and disadvantages. In calculation of the NMR chemical shift,the question on which level of theory is more applicable is under dispute.
2.2.2 Computation procedure
First, the geometric optimization of both ET and EG is calculated through CHEM 3D software. Second, ET and EG monomers, ET + 2H2O complex in solvation phase, EG +3H2O complex in solvation phase all use the various of basis sets at HF and DFT/B3LYP levels of theory to further optimize the geometrical structures, respectively. Third, the theoretical calculations of NMR chemical shift of EG-water mixtures adopt the GIAO method at HF and DFT levels of theory at different basis set. All computations are carried out using the Gaussian 09 program.
3 Results and discussion
In Fig.2,1H NMR spectra of the EG-water binary mixtures at 300.15 K at water volume fractions of ywater= 5%, 20% are depicted. It is observed that as the water content is small enough, the resonance absorption peak cannot be observed.With the increasing of the water content, the peak can be separated. Similar to this, at low concentration of alcohol the resonance peaks of the water proton and the alcoholic hydroxyl proton also cannot be separately observed. The reason for these phenomena is due to the fast proton exchanges. Only at the appropriate concentration of alcohol, the exchange of both protons in the mixtures is slow enough to individually be separated.
In Fig.3, the corrected chemical shifts of water protons and alcohol hydroxyl proton in the aqueous ET and EG mixtures at 300.15 K are plotted against the mole fraction of water,respectively. The chemical shift of water proton shows two different change trends in the ET-water mixture and the EG-water mixture. In ET solution, with the increasing water concentration, the water protons are rapidly shielded, and the resonance peaks swiftly shift to upfield, and the chemical shifts decrease dramatically. On the contrary, in EG solution, with the increasing water content, the water protons are moderately deshielded, and the resonance peaks shift to lowfield, and the chemical shifts increase slowly. The fact shows that the EG aqueous solutions have more “water-like” H-bond structures.One reason for the result is that EG has a hydroxyl group more than ET, and there is more H-bonds formed between the EG hydroxyl groups and the water protons in the EG-water mixtures. Moreover, Holmes et al.37observed that an increase in the basicity of a solvent increases the degree of covalence of the hydrogen bond between water and the organic solvent. And Forsyth et al.38thought that solvent basicity will affect the water proton chemical shift and the presence of the methyl group will results in a hydroxyl group more basic, which occurs as a result of the electron-donating ability of the methyl group which increases the electron density around the attached atom.Since there has a methyl group in ET molecular structure,which results in the hydroxyl group of ET more basic than that of EG, This feature makes the electron-donating ability of the methyl group of ET stronger, which can increase the electron density around the attached atom. This maybe another reason for the water proton chemical shift different changing trends in the ET and EG solutions. In both ET-H2O and EG-H2O solutions the water proton chemical shifts are higher than in pure water. In the case of EG-water mixtures, the water proton chemical shift is slightly higher than in pure water and reaches a maximum value at about 0.7 mole fraction of water. It is usually believed that the change of the water proton NMR chemical shift in a solution is the change of the intrinsic structure of water. The results obtained from Fig.3 indicate the rate of variation of water structure in ET solution is larger than in EG solution.
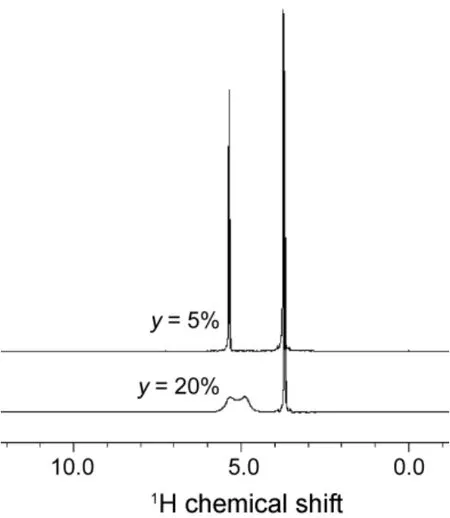
Fig.2 1H NMR spectra of EG-water mixtures as a function of water volume fraction, ywater = 5%, 20% at T = 300.15 K.

Fig.3 Experimental proton chemical shift of H2O protons and hydroxyl protons in ET and EG solutions relative to an external TMS reference. (at T = 300.15 K).
If there is a strong hydrogen-bond interaction between the alcohol oxygen and water protons, the alcohol proton will be deshielded and thus the peak will shift to lower field compared to the pure alcohol. From Fig.3, it is observed that with the increasing alcohol concentration the hydroxyl proton chemical shifts of ET and EG solution increase, which indicates the intermolecule H-bond formation and strengthening in alcohol aqueous solutions. It is considered that both the absolute values and the rate of change of chemical shift with increasing water content are dependent on the nature of the solute, it is necessary to compare the features of the chemical shift. It is interested to notice that in EG-water mixtures, the proton chemical shifts of both water and EG hydroxyl groups are not changed significantly with the water concentration. That is to say the EG alcohol resonances have only a slight dependence on concentration. While the rate of change of chemical shift of ET hydroxyl groups is greater than that of EG hydroxyl. This shows that stronger hydrogen-bonding interactions exist in ET-water mixtures than in EG-water mixtures. In contrast, in ET-water mixtures the water proton chemical shift is approximately 0.156 lower at initial the limiting chemical shift than that of the pure water. As the water concentration increases,the water proton chemical shift decreases and approaches 4.8(chemical shift of pure water). Compared to the EG aqueous solution, the ET aqueous solution has higher hydroxyl proton chemical shift at the same concentration. The difference is due to the solute different chemical structure. The higher electron density the O in ―OH groups has, the stronger the H-bond formed by the O atom is. The stronger H-bond formed makes the hydroxyl proton resonance peak shift to lower field in ET-H2O mixture, which make that the ―OH proton chemical shift value is larger. The result obtained in the paper is in agreement with that of the literature38. Furthermore, it is found that hydroxyl proton resonance peak of ET and EG shifts to lower field with decreasing water concentration. The low-field shifts may be explained in terms of the increase in both the polarization of the O―H bond and the H-bonding network strength of the hydroxyl hydrogen with increasing solute concentration.
Fig.4 and Fig.5 show mole fraction dependence of1H chemical shift of the alkyl proton of ET and EG in alcohol and water mixtures at different temperature, respectively. It is significantly noticed that the alkyl protons shift to upfield much more than the hydroxyl proton. Almost all alkyl protons show the same properties. That is the resonance peaks of all alkyl proton shift monotonically to low field with increasing alcohol concentration. As Mizuno et al.15interpreted, These results,shown in Fig.4 and Fig.5, are due to gradual electron transfer from the alkyl hydrogen to the oxygen with increasing alcohol concentration. The partial electron transfer of alkyl hydrogen and carbon to the oxygen of OH assists the formation of weak C―O…H H-bond in alcohol aqueous solutions. In general, that C―H groups can form weak C―O…H H-bond is accepted by many workers. Gu and Kar39indicate that the C―O…H interaction shows the similar features with conventional O―H…O H-bond in many respects in terms of the ab initio calculations..
The partial electron transfer creates almost linear increase of hydrogen chemical shift of alkyl groups. In ET aqueous solutions, the linear fitting results of the alkyl proton chemical shift show that the curve changes differently for different proton. For CH2, the curve slopes change from 0.512 to 0.549 with increasing temperature. While for CH3, the curve slopes just change from 0.498 to 0.509. Thus, for ET, the temperature dependence is CH3> CH2. As we know, the H-bond formation is dependent on the temperature. With increasing temperature,the H-bond tends to weaken and break. For CH3the sensitivity to temperature change is the higher, so it has the lower capability to forming H-bond. The result obtained is coincident with the PG aqueous solution3.
The literature40found that the excess molar enthalpy of all systems show eccentric behavior in the alcohol-water mixtures.Compared to the ideal solution, the mixtures reduce more negatively. Dixit et al.41found that the negative excess entropy observed in these system results from incomplete mixing at the molecular level, rather than from water restructuring. To better understand the “incomplete mixing” and anomalous polarization42of water in the mixtures, comparing the actual mixtures with the ideal mixtures is very necessary. The following relations were used to obtain the weighted average chemical shift of hydroxyl protons of alcohol and water in the actual mixtures δsol(X)3:


Fig.4 Mole fraction dependence of experimental 1H chemical shift of the alkyl proton of ET in ET-water mixtures at different temperature.(a) methylene(-CH2) proton; (b) methyl(-CH3) proton.

Fig.5 Mole fraction dependence of experimental 1H chemical shift of the alkyl proton of EG in EG-water mixtures at different temperature.
where δwater(X) represents the chemical shifts of the water proton; δalc(X) is the chemical shifts of alcohol hydroxyl proton.
Assuming an ideal alcohol-water mixtures with the chemical shift of the coalesced OH proton, the following formula is used to compute the weighted average of the chemical shifts for pure water and pure alcohol δsolΔ(X)3:

where δwaterand δalcare the chemical shifts for the hydroxyl protons of pure water and pure alcohol, respectively. Thus,δsolΔ(X) represents the chemical shift of an ideal mixture in which there is no interaction between solute and solvent. The solutions are the most “incomplete mixing”. δsolΔ(X) and δsol(X)for ET and EG are shown in Fig.6, respectively.
Δδ represents the difference between δsolΔ(X) and δsol(X). The bigger the value of Δδ, the more interaction of the solute and the solvent. As shown in Fig.3, the water proton chemical shifts of the ET and EG aqueous solutions are all much higher than the pure water chemical shift (about 4.8). The chemical shift of ET hydroxyl in solution is much lower than that of pure ET in the whole concentration region. On the contrary, the chemical shift of EG hydroxyl in solution is slightly lower than that of pure EG in the whole concentration region. On the whole, the degrees of water protons chemical shift changes in both ET and EG solution are larger than the hydroxyls protons. Therefore,The weighted average values δsol(X) of both ET and EG are higher than δsolΔ(X) in the whole region. For ET-water mixtures,the H-bond network is stronger in the range of 0.6−0.8 mole fraction of ET. While for EG-water mixtures, the H-bond network is stronger in the range of 0.0−0.3 mole fraction of EG.In the ET and EG aqueous solutions, the H-bond network is more stable than in the “incomplete mixing” solution.
4 Theoretical computation
4.1 Geometry of the ET-water complex and the EG-water complex
Raman et al.14has found that each glycerol molecule bonding with four water molecule forms a complex in a cluster organization irrespective of the glycerol content. In view of this,we will study the ET-water and EG-water interactions by treat water as continuum medium around the ET with two water molecules and the EG with three water molecules, respectively.There are controversies about the existence of H-bonds in short-chain alcohols/H2O mixtures. Mizuno et al.15Believed that there are four types hydrogen bonds in ET-water mixtures and the H-bond is formed between hydroxyl hydrogen and oxygen in mixtures. Zana and Eljebari43concluded that the self-associated aggregates of the short-chain alcohols are present in the aqueous solutions. Nishi et al.44thought H-bonding energy of ET-ET is nearly the same as that of H2O-ET in terms of ab initio calculations. while DʹArrigo and Teixeira45concluded that ET molecules are not associated with each other. However, the H-bonding formed between water molecules and alcohols molecules and the H-bonding formed between water molecules have been recognized by all scholars.So in this article, the H-bonding between water molecules and between water molecules and alcohol molecules have been taken into account. The optimized structures of ET and EG monomer in gas phase are shown in Fig.1. And the optimized structures of ET + 2H2O complex and EG + 3H2O complex in solvation phase are shown in Fig.7. The optimization geometrical parameters are enumerated in Tables 1−4 by use of the ab initio method GIAO at the different level.

Fig.6 Mole fraction dependence of 1H chemical shifts for δsol(X)and δsolΔ(X) in ET-water and EG-water mixture.(at T=300.15 K).(a) is for ET;(b) is for EG δsol(X)—the weighted average chemical shift of hydroxyl protons of alcohol and water in the actual mixtures;δsolΔ(X)—the chemical shift of an ideal mixture in which there is no interaction between solute and solvent.
The results obtained from Table 1−4 show that for ET-H2O complexes, the C―H bond length almost does not change compared to ET monomer geometry regardless of the method used. At HF/ 6-311 G++(d,p) level, the bond length ranges from 0.1085−0.1089 nm in ET monomer, and the bond length ranges from 0.1086 to 0.1087 nm in complex. And the maximum increase in C―H bond lengths in ET at HF/6-311 G++(d,p) level is just 0.0002 nm. At B3LYP/6-311 G++(d,p)level the range of the bond length is 0.1093−0.1098 nm in ET monomer, and the range of the bond length is 0.1096−0.1099 nm in complex. And the maximum increase in C―H bond lengths in ET at B3LYP/6-311 G++(d,p) level is just 0.0003 nm. While for O―H bond of ET, the bond length changes more.At HF/6-311 G++(d,p) and B3LYP/6-311 G++(d,p) level, the increases in O―H bond length of ET is 0.0028 nm and 0.0026 nm, respectively. On the other hand, for EG, though the bond length of C―H changes still small, the C―H bond length is larger in monomer than in complex. For O―H bond of EG, at HF/6-311 G(d) and B3LYP/6-311 G(d) level the maximum increases bond length are 0.002 and 0.0025 nm, respectively.The reason for this is due to the formation of H-bond between the hydroxyl groups of alcohol with water molecules and the weakening of O―H bonds in alcohols.

Fig.7 Optimized structural parameters of the ET + 2H2O complex and the EG + 3H2O in solvation phase.

Table 1 Optimized structural parameters of the ET monomer in gas phase calculated at HF/6-311 G++(d,p) and B3LYP/6-311 G++(d,p) level of theory.

Table 2 Optimized structural parameters of the ET + 2H2O complex in solvation phase calculated at HF/6-311 G++(d,p) and B3LYP/6-311 G++(d,p) level of theory.
It is observed from Table 2 and Table 4 that in solvation phase the O―H…O H-bond between the alcohol molecule and water molecule is formed. The O―H…O H-bonds include two styles in ET aqueous solutions: O(8)―H(9)ET… O(13)W,O(10)―H(11)W…O(8)ET. While in EG aqueous solutions there are four types: O(7)―H(8)EG…O(14)W, O(9)―H(10)EG…O(11)W, O(11)―H(12)W…O(7)EG, O(17)―H(18)W…O(9)EG. In addition, there is still H-bond between water molecules. These are O(13)―H(14)W…O(10)Wfor ET and O(14)―H(16)W…O(17)Wfor EG. For ET-H2O mixtures, the bond length of O―H…O H-bond are in the range of 0.2047−0.2414 nm at HF/6-311 G++(d,p) level and in the range of 0.1989−0.1998 nm at B3LYP/6-311 G++(d,p) level. For EG-H2O mixtures, the bond length of O―H…O H-bond varies between 0.1876–0.1982 nm at HF/6-311 G(d) level and between 0.1715–0.1802 nm at B3LYP/6-311G(d) level. These values of O―H…O H-bond bond length are much larger than those of C―C bond,C―H bond and O―H bond. The fact indicates that the O―H…O H-bond formed between alcohol molecules and water molecules are weaker than those of intramolecular H-bonds and other polar covalent bonds. By contrast, it is found that the O―H bond formed between the O in ET molecule and H in water molecule is slightly more stable than that formed between the H in ET molecule and O in water molecule. Compared with ET, EG does not show the same law.Furthermore, comparing the alcohol monomer with the alcohol-water complex solvation phase, it is found that the bond length values of C―C bond and C―O bond of ET and EG at different levels and by different methods both increase.The fact shows that the hydrogen bond interactions make the C―C and C―O bonds of alcohol weak to a different degree.Besides, the O―HW…OETbond angle is 170.512° and the O―HET…OWbond angle is 145.414° for ET solutions. For EG,the O―HW…OEGbond angle varies in the range of 171.142°and 175.918° and the O―HEG…OWbond angle is in the range of 157.347°−166.872°.
It is interesting to notice that the bond length values computed of C―C, C―H and O―H bond of both ET and EG are larger at DFT(B3LYP) level than at HF level of theory at the same polarization function and diffusion function. For example, for EG-H2O complex in solvation phase, the C(2)―H(5) bond length at HF/6-311G++(d,p) level of theory is 0.1085 nm. While the C(2)―H(5) bond length at B3LYP/6-311G++(d,p) level of theory is 0.1096 nm. By computation, it is found that the maximum increasing percentages are 2%, 4%,2% and 3% for ET monomer, ET-water mixtures, EG monomer and EG-water mixtures, respectively. The reason for that result is due to the electronic self-interaction at DFT(B3LYP) level of theory. The self-interaction makes the structure more loose. On the contrary, the O―H…O H-bond bond length values computed at HF/6-311 G++(d,p) level are larger than those at B3LYP/6-311G++(d,p) level for both ET-water mixtures and EG-water mixtures. For ET, the maximum percentage of reduction is 6.7%. While for EG, the maximum percentage of reduction even reached 9.1%. The fact means that the O―H…O H-bond obtained by DFT level of theory is stronger than by HF level of theory. In addition, the computing results of Table 2 and Table 4 show that adding polarization function and diffusion function to the basis set at the same level of theory can improve the bond length of alcohol.

Table 3 Optimized structural parameters of the EG monomer in gas phase calculated at HF/6-311 G(d) and B3LYP/6-311 G(d) level of theory

Table 4 Optimized structural parameters of the EG +3H2O complex in solvation phase calculated at HF/ 6-311 G(d)and B3LYP/ 6-311 G(d) level of theory
4.2 Theroretical Calculation for NMR
In EG-water mixtures, the theoretical calculation of NMR is based on Hartree-Fork and DFT levels of theory. In geometric optimization, the DFT(B3LYP)/6-311G++(d,p) basis set is used. In this paper, we take GIAO method to calculate NMR for EG-water mixtures. In Table 5, the results of1H NMR chemical shift computed are shown. It is observed that as far as the theory levels are concerned, DFT(B3LYP) is usually better than HF. The computing accuracy of DFT(B3LYP) is higher than HF. The advantage of DFT is to take into account the electron correlation energy. So the computational accuracy of DFT(B3LYP) level of theory is higher.
As we know, the calculation accuracy can be improved by adding polarization function and diffusion function to the basis set. From Table 5, with the polarization function and diffusion function increasing, the results of calculation approach the experimental values. This shows that as for the same method,the larger the basis sets are, the more accurate the calculated values are. As stated previously, in geometric optimization the value of O―H…O bond length at HF level of theory is larger than at DFT level of theory. The fact indicates that the O―H…O H-bond at HF level of theory is stronger than at DFT level of theory. The stronger the O―H…O H-bond is, the greater the deshielding effect is. And the absorption peak shifts downfield.Thus, the1H chemical shift increases. In Table 5, the computing values are in good agreement with the results of geometrical optimization. Fig.8 shows the experimental and the theoretical1H chemical shift of hydroxyl proton in the aqueous EG mixtures at 300.15 K vs EG mole fractions. It is observed that on the whole fraction scale the accuracy of DFT(B3LYP)level of theory is higher than that of HF/6-311G++(d,p). The maximum deviation of HF/6-311G++(d,p) is 5.1%. While the maximum deviation of DFT(B3LYP)/6-311G++(d,p) is just 0.37%.

Table 5 1H NMR chemical shift theoretical computation data of EG aqueous solution at different theory levels (Opt B3LYP/ 6-311 G++(d,p))

Fig. 8 Experimental and theoretic chemical shift of hydroxyl proton in the EG-water mixtures at T = 300.15 K.
5 Conclusions
To better understand the interaction between water and alcohol with two carbon atoms,1H NMR experiment measurement and quantum chemistry theoretical computation have been conducted on both mixtures. The conclusions obtained in this paper are as follows:
(1) The experimental proton chemical shifts of water and double-carbon alcohol in alcohol-water complex indicate that in alcohol-water mixtures the H-bond does exist. And the two mixtures show distinct changing trend in the chemical shifts of water protons and alcohol hydroxyl protons. While for the chemical shifts of alkyl protons the two small molecular alcohols show the same changing trend.
(2) In many levels of theory of quantum chemistry, this paper chooses Hartree-Fork and density functional theory to optimize geometrical and compute NMR magnetic shielding constants.The geometrical structure optimization parameters at different basis sets show that for alcohol-H2O complexes in solvation phase, the C―H bond length almost does not change compared to alcohol monomer geometry regardless of the levels used.While for O―H bond of alcohol, the bond lengths change more.The reason for this is due to the formation of H-bond between the hydroxyl groups of alcohol with water molecules and the weakening of O―H bonds in alcohols. By comparison, it is found that C―C, C―O and C―H bond length obtained by the DFT theory is longer than by the HF theory. The reason for this is that the structure obtained by DFT theory is more loose.However, the O―H…O bond length obtained by the DFT theory is shorter than by the HF theory. The reason is due to the neglect of electron correlation in HF level of theory, which makes HF not be good at deal with the weak bond, such as O―H…O H-bond.
(3) In prediction of NMR chemical shift, as far as the theory levels are concerned, DFT(B3LYP) is usually better than HF.As for the same methods, the larger the basis sets are, the more accurate the calculated values are.
(4) The experimental results are in good agreement with the theoretical results. By DFT(B3LYP)/6-311G++(d,p), the prediction of NMR chemical shift is more accurate. Thus in future, the NMR chemical shift computation of other alcohols will use the above method, level of theory and basis set to obtain the better values.
Acknowledgment: We thank the analysis and testing center of Anhui University for its support in the process of experiment and calculating.
(1) Raman, M. S.; Ponnuswamy, V.; Kolandaivel, P.; Perumal, K. J. Mol.Liq. 2007, 135 (1−3), 46. doi: 10.1016/j.molliq.2006.10.011.
(2) Fang, Y. J.; Zhou, P. Sep. Sci. Technol. 2006, 41 (2), 329.doi: 10.1080/01496390500460666.
(3) Zhou, Y.; Hu, K.; Shen, J.; Wu, X.; Cheng, G. J. Mol. Struct. 2009,921 (1), 150. doi: 10.1016/j.molstruc.2008.12.050.
(4) Chen, C.; Li, W. Z. Acta Phys. -Chim. Sin. 2009, 25 (3), 507. [陈 聪,李维仲. 物理化学学报, 2009, 25 (3), 507.]doi: 10.3866/PKU.WHXB20090318
(5) Zhao, J. F.; Sun, X. L.; Li, J. L.; Huang, X. R. Acta Phys. -Chim. Sin.2015, 31 (6), 1077. [赵俊凤, 孙小丽, 李吉来, 黄旭日. 物理化学学报, 2015, 31 (6), 1077.] doi: 10.3866/PKU.WHXB201504014
(6) Gao, C.; Wang, T. J.; Liu, X. N.; Zhou, G. Y.; Hua, T. C. Chin. J.Chem. Phys. 2007, 20 (3), 258.
(7) Ye, B.; Gao, C.; Liu, X. N.; Yang, S.; Jiang, B. Acta Phys. -Chim. Sin.2011, 27 (5), 1031. [叶 斌, 高 才, 刘向农, 杨 锁, 江 斌.物理化学学报, 2011, 27 (5), 1031.]doi: 10.3866/PKU.WHXB20110419
(8) Sengwa, R. J.; Sankhla, S.; Shinyashiki, N. J. Phys. Chem. 2008, 94(17), 6889. doi: 10.1007/s10953-007-9230-6
(9) Ye, B.; Gao, C.; Zhao, H.; Chen, K. S.; Yang, S.; Liu, X. N. Acta Phys. -Chim. Sin. 2011, 27 (11), 2505. [叶 斌, 高 才, 赵 韩,陈开松, 杨 锁, 刘向农. 物理化学学报, 2011, 27 (11), 2505.]doi: 10.3866/PKU.WHXB20111103
(10) Zhang, N.; Li, W. Z.; Chen, C.; Zuo, J.G.; Weng, L. D. Bull.Korean Chem. Soc. 2013, 34 (9), 2711.doi: 10.5012/bkcs.2013.34.9.2711
(11) Zhang, N.; Li, W. Z.; Chen, C.; Zuo, J.G.; Weng, L. D. Mol.Phys. 2013, 111 (7), 939. doi: 10.1080/00268976.2012.760050
(12) Chen, C.; Li, W. Z.; Song,Y. C.; Weng, L. D.; Zhang, N. Mol.Phys. 2012, 110 (5), 283. doi: 10.1080/00268976.2011.641602
(13) Lee, M.; Hong, M. J. Biomol. NMR 2014, 59 (4), 263.doi: 10.1007/s10858-014-9845-z
(14) Raman, M. S.; Ponnuswamy,V.; Kolandaivel, P.; Perumal, K.J. Mol. Liq. 2008, 142 (1−3), 10.doi: 10.1016/j.molliq.2008.03.006
(15) Mizuno, K.; Miyashita, Y.; Shindo, Y.; Ogawa, H. J. Phys.Chem. 1995, 99 (10), 3225. doi: 10.1021/j100010a037
(16) Zhang, S.G.; Yang, P. J. Chin. Soc. Corros. Prot. 2004, 24 (4),240. [张士国, 杨 频. 中国腐蚀和防护学报, 2004, 24 (4),240.]
(17) Zarzycki, P.; Rustad, J. R. J. Phys. Chem. A 2009, 113 (1), 291.doi: 10.1021/jp805737a
(18) Lü sse, S.; Arnold, K. Macromolecules 1996, 29 (12), 4251.doi: 10.1021/ma9508616
(19) Song, B. T.; Chu, Y. Y.; Wang, J. Q.; Zheng, A. M.; Deng, F.Chin. J. Magn. Reson. 2016, 33 (3), 378. [宋本腾, 褚月英,王吉清, 郑安民, 邓风. 波谱学杂志, 2016, 33 (3), 378.]doi: 10.11938/cjmr20160303
(20) Li, W.; Zhang, J.; Qi, C. S. Acta Phys. -Chim. Sin. 2015, 31 (9),1690. [李 巍, 张 静, 戚传松. 物理化学学报, 2015, 31(9), 1690.] doi: 10.3866/PKU.WHXB201507071
(21) Zhang, X. X; Chen, Q. C; Hu, W. H.; Zhang, J. B. Appl. Surf.Sci. 2013, 286 (12), 47. doi: 10.1016/j.apsusc.2013.09.005
(22) Lan, R.; Li, H. R.; Han, S. J. Acta Phys. -Chim. Sin. 2005, 21(11), 1295. [蓝 蓉, 李浩然, 韩世钧. 物理化学学报, 2005,21 (11), 1295.] doi: 10.3866/PKU.WHXB20051120
(23) Liao, L. M.; Huang, X.; Li, J. F. Chin. J. Magn. Reson. 2016,33 (3), 368. [廖立敏, 黄 茜, 李建凤. 波谱学杂志, 2016,33 (3), 368.] doi: 10.11938/cjmr20160302
(24) Wang, Y. H.; Li, X. Y.; Zeng, Y. L.; Meng, L. P.; Zhang, X. Y.Acta Phys. -Chim. Sin. 2016, 32 (3), 671.[王月红, 李晓艳,曾艳丽, 孟令鹏, 张雪英. 物理化学学报, 2016, 32 (3), 671.]doi: 10.3866/PKU.WHXB 201512293
(25) Xu, X.; Xu, Z. G.; Luo, Y. F. Acta Phys. -Chim. Sin. 2002, 18(5), 420. [许 旋, 徐志广, 罗一帆. 物理化学学报, 2002,18 (5), 420.] doi: 10.3866/PKU.WHXB20020508
(26) Wang, Y. N.; Jin, Q.; Li, H. D.; Wang, C. J. Chin. J. Magn.Reson. 2015, 32 (3), 528. [王燕妮, 金 芩, 李慧丹, 王朝杰.波谱学杂志, 2015, 32 (3), 528.] doi: 10.11938/cjmr20150314
(27) Mizuno, K.; Kimura, Y.; Morichika, H.; Nishimura, Y.;Shimada, S.; Maeda, S.; Imafuji, S.; Ochi, T. J. Mol. Liq. 2000,85 (1−2), 139. doi: 10.1016/S0167-7322(99)00170-1
(28) Covington, A. K.; Newman, K. E. Modern Aspects of Electrochemistry; Bockris, J. OM.; Conway, B. E.; Eds.;Pergamon: Oxford, U.K.; 1977; Vol. 12, pp 41−129.
(29) Pople, J. A.; Schneider, W. G.; Barnstein, H. J. High Resolution NMR, 2nd ed.; McGraw-Hill: New York, 1967;pp 16−136.
(30) Selwood, P. W. Magnetochemistry,1nd ed.; Interscience: New York, 1943; pp 12−129.
(31) Hinton, J. F.; Wolinski, K. Ab initio GIAO Magnetic Shielding Tensor for Hydrogen-bonded Systems. In Theoretical Treatments of Hydrogen Bonding; Hadzi, D.; Ed.;Wiley: Chichester, U.K.; 1997; p 75.
(32) Chesnut, D. B. The Ab Initio Computation of Nuclear Magnetic Resonance Chemical shielding. In Reviews in Computational Chemistry; Lepkowitz, K. B., Boyd, D. B.,Eds.; VCH Publishers: New York, 1996; Vol. 8, pp 245−297.
(33) Fleischer, U.; van Wü llen, C.; Kutzelnigg, W. NMR Chemical Shift Computation: Ab Initio in Encyclopedia of Computational Chemistry; Wiley: New York, 1998; Vol. 3,pp 35−67.
(34) Cheeseman, J. R.; Trucks, G. W.; Keith, T. A.; Frisch, M. J.J. Chem. Phys. 1996, 104 (14), 5497. doi: 10.1063/1.471789
(35) Hohenberg, P.; Kohn, W. Phys. Rev. B 1964, 136 (3), 864.doi: 10.1103/PhysRev.136.B864
(36) Kohn, W.; Sham, L. J. Phys. Rev. A 1965, 137 (6A), 1697.doi: 10.1103/PhysRev.137.A1697
(37) Holmes, J. R.; Kivelson, D.; Drinkard, W. C. J. Am. Chem.Soc. 1962, 84 (24), 4677. doi: 10.1021/ja00883a013
(38) Forsyth, M.; Macfarlane, D. R. J. Phys. Chem. 1990, 94 (17),6889. doi: 10.1021/j100380a064
(39) Gu, Y.; Kar, T.; Scheiner, S. J. Am. Chem. Soc. 1999, 121 (40),9411. doi: 10.1021/Ja991795g
(40) Nagamachi, M. Y.; Francesconi, A. Z. J. Chem.Thermodynamics 2006, 38 (4), 461.doi: 10.1016/j.jct.2005.06.018
(41) Dixit, S.; Crain, J.; Poon, W. C.; Finney, J. L.; Soper, A. K.Nature 2002, 416 (6883), 829. doi: 10.1038/416829a
(42) Mizuno, K.; Imafuji,S.; Fujiwara,T.; Ohta, T.; Tamiya,Y.J. Phys. Chem. B. 2003, 107 (16), 3972.doi: 10.1021/jp021712+
(43) Zana, R.; Eljebari, M. J. J. Phys. Chem. 1993, 97 (42), 11134.doi: 10.1021/j100144a039
(44) Nishi, N.; Koga, K.; Ohshima, C.; Yamamoto, K.; Nagashima,U.; Nagami, K. J. Am. Chem. Soc. 1988, 110 (16), 5246.doi: 10.1021/ja00224a002
(45) Dʹ Arrigo, G.; Teixeira, J. J. Chem. Soc. Faraday Trans. 1990,86 (9), 1503. doi: 10.1039/FT9908601503
Experimental and Theoretical Analysis of1H NMR on Double-Carbon Alcohol Aqueous Solutions
YE Bin1ZHANG Jian2GAO Cai1,*TANG Jing-Chun1
(1School of Automobile and Transportation Engineering, Hefei University of Technology, Hefei 230009, P. R. China;2School of Electrical Engineering and Automation, Hefei University of Technology, Hefei 230009, P. R. China)
In this study,1H nuclear magnetic resonance (NMR) measurements and quantum chemistry(QC) studies of ethanol (ET)-water mixtures and ethylene glycol (EG)-water mixtures are carried out at different temperatures to discuss the interactions between water and the alcohols present in the mixtures.From1H NMR spectra, it is observed that the chemical shift of the water proton shows two different trends in the ET-water mixtures and the EG-water mixtures. With increasing water concentration, the water proton chemical shift decreases dramatically for ET-water mixtures, while the chemical shift increases slowly for EG-water mixtures. The alcohol hydroxyl proton resonance peaks of both ET and EG shift to lower field with decreasing water concentration. It is found that the resonance peaks of all alkyl protons shift monotonically to low field with increasing alcohol concentration at different temperatures. The geometry optimization results indicate the formation of H-bonds between the water molecules and the hydroxyl groups of the alcohols alongside the weakening of O―H bonds in the alcohols, which results in an O―H bond length decrease. It is interesting to note that the bond length values computed for C―C,C―H and O―H bond in both ET and EG are larger when calculated at the density functional theory (DFT)(B3LYP) level than when calculated using Hartree-Fock (HF) level of theory with the samepolarization function and diffusion function. However, the O―H···O H-bond computed at HF level of theory is stronger than that calculated at DFT level of theory. The theoretical results are in good agreement with the experimental ones. In the calculation of NMR chemical shift, DFT(B3LYP) is better than HF, which implies that for the same method, the larger the basis sets are, the more accurate are the calculated values.
Ethanol; Ethylene glycol;1H Nuclear magnetic resonance; Chemical shift;Hartree-Fork level of theory; Density functional theory
April 6, 2017; Revised: May 2, 2017; Published online: May 12, 2017.*
. Email: gao_cai@hotmail.com; Tel: +86-13721029540.
© Editorial office of Acta Physico-Chimica Sinica
O641
10.3866/PKU.WHXB201705124 www.whxb.pku.edu.cn
猜你喜欢
波谱学杂志(2021年3期)2021-09-07
合肥工业大学学报(社会科学版)(2020年2期)2020-05-14
合肥工业大学学报(自然科学版)(2020年1期)2020-02-24
山西教育·招考(2019年12期)2019-09-10
山西教育·招考(2019年12期)2019-09-10
山西教育·招考(2019年12期)2019-09-10
物理化学学报(2018年6期)2018-03-08
浙江工业大学学报(2017年5期)2018-01-22
校园英语·中旬(2017年11期)2017-10-25
印制电路信息(2015年6期)2015-12-30
