
2,4-二硫代尿嘧啶的紫外吸收光谱和共振拉曼光谱
2017-11-01 18:11金颖淳郑旭明
物理化学学报 2017年10期
金颖淳 郑旭明
(浙江理工大学化学系,杭州 310018)
2,4-二硫代尿嘧啶的紫外吸收光谱和共振拉曼光谱
金颖淳 郑旭明*
(浙江理工大学化学系,杭州 310018)
硫代嘧啶碱基是光动力疗法潜在的重要光敏剂,其最低单重激发态的光物理研究已有广泛报道。然而,其较高激发态的跃迁性质和反应动力学研究较为稀少。因此,本文采用共振拉曼光谱和密度泛函理论计算方法研究2,4-二硫代尿嘧啶的紫外光谱和几个较高单重激发态的短时结构动力学。首先,基于共振拉曼光谱强度与电子吸收带振子强度f的关系,将紫外光谱去卷积成四个吸收带,分别为358 nm (f =0.0336)中等强度吸收带(A带),338 nm (f = 0.1491)、301 nm (f = 0.1795)和278 nm (f = 0.3532)强而宽的吸收带(B、C和D带)。这一结果既吻合密度泛函理论计算结果,又符合共振拉曼光谱强度模式对紫外光谱带的预期。据此,去卷积得到的四个吸收带被分别指认为S0→ S2跃迁、S0→ S6跃迁、S0→ S7跃迁和S0→ S8跃迁。同时,分别对B,C和D带共振拉曼光谱进行了详细的指认,获得了短时动力学信息。结果表明,S8态短时动力学的显著特征是在Franck-Condon区域或附近发生了S8(ππ*)/S(nπ*)势能面交叉引发的、伴随超快结构扭转的非绝热过程。S7和S6态短时动力学的主要特征是反应坐标的多维性,它们分别沿C5C6/C2S8/C4S10/N2C3+ C4N3H9/N1C2N3/C2N1C6/C6N1H7/C5C6H12和C5C6/N3C2/C4S10/C2S8+C6N1H7/C5C6H12/C5C6N1/C5C6H12/C2N1C6/N1C2N3/C4N3H9/N1C2N3等内坐标演化。
2,4-二硫代尿嘧啶;激发态结构动力学;紫外吸收光谱;共振拉曼光谱;密度泛函理论
1 引 言
天然核酸碱基受光诱导的激发态衰变动力学是一个由 S2(ππ*)/S0势能面交叉主导的超快内转换过程1–5。这一过程能使激发态分子来不及发生化学反应就回到了基态,从而保障了生命体中的DNA分子免受紫外辐射影响,使遗传信息复录得以正常进行。与天然核酸碱基不同,硫代核酸碱基的低能态激发态衰变过程主要受系间窜越过程的控制6–16。例如,2-硫代胸腺嘧啶初始布居单重态的主要弛豫通道为系间窜越到 T1(ππ*)6–9,2-硫代胸腺嘧啶6,8,9、4-硫代胸腺嘧啶8,10–13和硫代鸟嘌呤14等分子经系间窜越到到T1(ππ*)态的量子产率很高,有些接近1。硫代碱基具有较高的光敏化单线态氧产率,具体与结构有关8。这使得它们在光动力疗法等领域倍受关注。
在动力学方面,纳秒、皮秒和飞秒时间分辨瞬态吸收光谱技术揭示了2-硫代胸腺嘧啶T1(ππ*)形成的时间尺度9,11–13。理论计算研究洞察了硫代尿嘧啶和硫代鸟嘌呤系间窜越过程的微观机制16,17。借助于全活化空间自洽场(CASSCF)理论及其二阶微扰(CASPT2)方法所获得的2-硫代尿嘧啶激发态势能面和旋-轨耦合信息,崔刚龙和方维海提出了从初始布居态 S2(ππ*)产生最低三重态 T1(ππ*)的三条互相竞争、高效的非绝热通道17。Pollum and Crespo-Hernández18采用飞秒时间分辨宽带瞬态吸收实验评估了崔刚龙和方维海提出的三条ISC 通道,提出了 S2(ππ*) → S1(nπ*) → T1(ππ*)过程是主要通道。这一结论与前期Gobbo的研究结果一致19。目前认为,S1(nπ*)态是完成 S2(ππ*) →T1(ππ*)系间窜越的门态,决定着 T1(ππ*)量子产率大小20,21。姜杰等22采用共振拉曼光谱结合CASSCF方法研究了 2-硫代尿嘧啶激发态衰变动力学,揭示经 S2(ππ*)/S1(nπ*)势能面交叉从 S2(ππ*)态弛豫到S1(nπ*)态是产生T1(ππ*)态的主要通道。最近,崔刚龙等采用 CASPT2//CASSCF和QM(CASPT2// CASSCF)/MM方法提出了2,4-二硫代胸腺嘧啶(2,4-diiothymine)在真空、微溶剂化和水环境下的激发态衰变机制和溶剂效应23。
2,4-二硫代尿嘧啶(简称 24DTU)是尿嘧啶分子中两个羰基氧原子均被硫原子取代的产物。相对于单硫代碱基,二硫代碱基的电子态结构较为复杂,紫外光区多个光吸收态的存在使得紫外光谱指认变得不确定。因此,本文拟借助共振拉曼光谱的强度模式对不同电子态的对应关系,通过紫外吸收光谱和共振拉曼光谱实验,对比密度泛函理论计算结果,研究24DTU分子激发态电子结构,指认紫外吸收光谱和共振拉曼光谱,以获得电子态耦合信息和短时结构动力学结果。
2 实验部分
2.1 试剂和仪器
24DTU (2,4-dithiouracil) (99.9%,北京百灵威科技有限公司),乙腈(99.9%,斯百全化学有限公司,甲醇(99.9%,斯百全化学有限公司,纯水(杭州娃哈哈集团有限公司)。
紫外可见分光光度计(Cary 50,瓦里安公司,美国),傅里叶红外光谱仪(Nicolet Avatar 370,尼高力公司,美国);傅里叶拉曼光谱仪(Nicolet Raman 960,尼高力公司,美国);共振拉曼光谱仪(自制)。
2.2 实验方法
24DTU在乙腈、甲醇和水中的浓度约为0.90–1.0 × 10−3mol·L−1。共振拉曼光谱实验装置和方法参见文献24。24DTU的共振拉曼光谱的激发波长(266.0、273.9、282.4、299.1、309.1、319.9、341.5和354.7 nm)由10 Hz Nd:YAG激光器的二、三、四倍频激光(266.0、355和532 nm)和它们的氢受激拉曼线提供。样品溶液由循环泵输送至喷嘴口,喷嘴口可以挤压溶液使得通过的溶液变成薄膜状。此时,通过棱镜折射后的激光打在溶液薄膜上,椭球镜则收集拉曼散射的信号,最后Princeton Instruments PyLoN:2KBUV型CCD检测器(2048 ×512前感光液氮冷却)收集信号并将它们显示在计算机上。喷嘴口流出的溶液则通过漏斗收集,由导管通入进锥形瓶,产生溶液流动的循环。共振拉曼光谱的一次采集时间由强度或整体信噪比决定,一般在100–150 s。50–100次采集数据的累加结果即为该激发波长下的共振拉曼光谱原图。原图经像素-波数转换(cm−1)、灵敏度和自吸收等校正、溶剂峰扣除等处理后得到样品的共振拉曼光谱图。

图1 (a) 24DTU在乙腈、甲醇和水中的紫外吸收光谱;(b) 在PCM溶剂模型下由B3LYP/6-31+G(d)计算获得的24DTU的几何结构示意图Fig.1 (a) UV absorption spectra of 24DTU in acetonitrile, methanol and water; (b) Schematic diagram of the geometry structure of 24DTU.
3 理论计算
电子基态几何结构和振动频率采用密度泛函理论在极化连续介质模型(PCM)溶剂模型和B3LYP/6-31+G(d)计算水平下计算获得。电子跃迁能和振子强度在 B3LYP-TD/6-311++G(3df,3pd)水平下计算获得。本文所有量子化学计算均采用Gaussian 09W程序包完成25。简正模式分析采用VEDA4程序完成26。
4 结果与讨论
4.1 紫外光谱研究
图1示出24DTU在乙腈、甲醇和水溶液中的紫外吸收光谱,图中箭头上方数字为共振拉曼光谱实验所用的激光波长。24DTU紫外光谱展示两个宽吸收带,分别位于282和346 nm,其中346 nm较强吸收带的不对称性预示该带由某些强吸收带组合。表 1列出 B3LYP-TD/6-311++G(3df,3pd)计算水平和PCM溶剂模型下获得的24DTU在乙腈中的电子跃迁能、跃迁轨道和振子强度。表 1可见,在 > 240 nm光谱区域,计算结果给出四个允许跃迁,最大吸收波长和振子强度f分别为336 nm(f = 0.0451),297 nm (f = 0.1472),272 nm (f =0.3851)和253 nm (f = 0.4066)。图2为24DTU在乙腈溶液中的紫外吸收光谱的去卷积图。图 2和表 1可见,去卷积获得的四个主要吸收峰的最大吸收波长和振子强度 f与理论计算结果又较好的相关性。因此,根据图1、图2和表1的结果,图2中的358 nm (f = 0.0336)中等强度吸收带(A带)被指认为S0→ S2跃迁, 338 nm (f = 0.1491)、301 nm (f = 0.1795)和278 nm (f = 0.3532)强而宽的吸收带(B、C 和 D 带)被分别指认为 S0→ S6、S0→ S7和S0→ S8跃迁。
值得指出的是,为了排除去卷积过程存在的随意性,综合考虑了不同激发波长下的共振拉曼光谱与振子强度的关系,即拉曼光谱强度与振子强度f的平方成正比。因此,把338 nm去卷积带(f =0.1491)指认为S0→ S6吸收带(f = 0.1789)而非S0→S2吸收带(f = 0.0541)。这得到了341.5和354.7 nm共振拉曼光谱强度模式的有力支持。此外,我们注意到,构成S0→ S6、S0→ S7和S0→ S7跃迁的主要轨道基本相同,区别在于相应的跃迁轨道系数大小或符号不同,这表明S6、S7、和S8三个态之间存在态态耦合。溶剂对紫外吸收光谱的结构和带宽影响不大。根据朗伯比尔定律(Lambert-Beer)定律,24DTU在乙腈、甲醇和水溶液中的最大吸收波长处的摩尔消光系数εmax/λmax分别为:2.29 × 104L·mol−1·cm−1(280.0 nm)、2.21 × 104L·mol−1·cm-1(283.0 nm)和2.20 × 104L·mol−1·cm−1(281.0 nm)。
4.2 振动光谱指认
图3示出24DTU的傅里叶变换拉曼和红外光谱,以及由B3LYP/6-31+G(d)计算获得并由GaussView软件输出的拉曼光谱(参数为半峰宽2 cm−1,步长1 cm−1)27。图3中,计算拉曼光谱频率已按实验光谱频率做了校正。表2列出实验和计算振动频率。表2可见,24DTU有30个振动模,其中属于A′和A′不可约表示的振动模分别有21个和9个。图3和表2可见,拉曼光谱中主要谱带的实验拉曼位移(频率)与计算值基本吻合,表明密度泛函理论计算结果总体合理。例如,实验拉曼光谱中的1549、1491、1253、1228、1188、1118、1077、683、460、443和229 cm−1等谱带与计算光谱中的ν6、ν7、ν10、ν11、ν12、ν13、ν14、ν17、ν18、ν19、ν21等有良好的一一对应关系。基于密度泛函理论计算结果开展了振动光谱的简正模式分析,获得的势能分布(potential energy distribution)列于表2。表2可见,除NH/CH伸缩振动外,绝大多数振动模的振动形式都由多个内坐标的复合运动构成。

图2 24DTU在乙腈溶液中的紫外吸收光谱的去卷积曲线(点线)Fig.2 Deconvoluted curves (dotted line) of the UV absorption spectrum of 24DTU in acetonitrile.

表1 在PCM溶剂模型下由B3LYP-TD/6-311++G(3df,3pd)计算获得的24DTU的电子跃迁能(△E)、跃迁轨道和振子强度(f)Table 1 Electronic transition energies (△E), transition orbitals and Oscillator strengths (f) for 24DTU computed by B3LYP-TD/6-311++G(3df,3pd) and polarization continum model (PCM).
4.3 共振拉曼光谱和短时结构动力学
图 4示出 266.0、273.9、282.4、299.1、309.1、319.9、341.5和 354.7 nm 激发下 500–1800 cm−1光谱区域内的共振拉曼光谱,以展现基频信息。

图3 24DTU的傅里叶变换红外和拉曼光谱以及B3LYP/6-31+G(d)计算拉曼光谱Fig.3 Comparison of FT-IR, FT-Raman and B3LYP/6-31+G(d) computed Raman spectra of 24DTU.

表2 24DTU在B3LYP/6-31+G(d)计算水平下的计算频率和实验观察到的傅里叶变换红外和拉曼光谱的振动频率和指认Table 2 Vibrational frequencies and assignments of 24DTU calculated by using B3LYP/6-31+G(d) and PCM model and observed by experimental FT-IR and FT-Raman spectra.
图5展示 500–3500 cm−1光谱区域内的266.0、319.9和341.5 nm共振拉曼光谱,以全面展现倍频和组合频信息。借助图3和表2,图4和图5中的共振拉曼光谱可被初步指认为由 ν17、ν16、ν15、ν14、ν13、ν11、ν9、ν8、ν7、ν6和 ν5等 11 个振动基频以及它们的倍频和组合频构成。为了排除因溶剂扣减引起的瑕疵峰对共振拉曼光谱的干扰和考察溶剂性质对24DTU共振拉曼光谱的强度模式影响,获取了在乙腈、甲醇和水中的共振拉曼光谱。图4可见,溶剂扣减的瑕疵峰和溶剂性质对共振拉曼光谱影响不大。
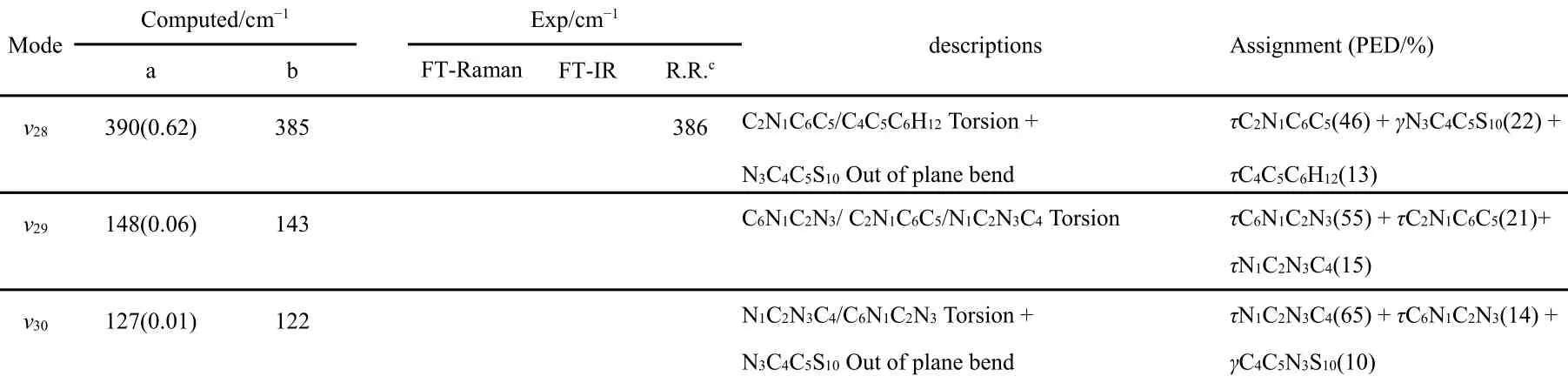
continued Table 2

图4 2.4-二硫代尿嘧啶在乙腈(a)、甲醇(b)和水(c)溶剂中的不同激发波长下的共振拉曼光谱Fig.4 Resonance Raman spectra of 24DTU in acetonitrile (a), methanol (b) and water (c) solvents.
图 4可见,在乙腈和甲醇中不同激发波长下的共振拉曼光谱存在良好的一一对应关系,表明24DTU的 B、C和 D带吸收跃迁的能量和Franck-Condon区域势能面构造在两种溶剂环境中基本相同。但水对24DTU的C吸收带产生明显的蓝移作用,并且相应的共振拉曼光谱强度模式与在乙腈和甲醇中的有一定的差异,表明C吸收带的跃迁能和对应势能面在 Franck-Condon区域的构造在水环境中发生了明显变化。
众所周知,共振拉曼光谱的强度模式对激发态性质很敏感。激发态性质不同,所对应的共振拉曼光谱的强度模式往往也不同。因此,通过改变激发波长使之与不同激发态共振,能获得不同激发态上的短时结构动力学信息。以水中共振拉曼光谱为例,图4(c)中341.5和354.7 nm共振拉曼光谱的强度模式基本相同,266.0、273.9和282.4 nm共振拉曼光谱的强度模式基本相同,而 309.1和319.9 nm共振拉曼光谱的强度模式基本相同,但与前两者都不同。显然,341.5和354.7 nm激发波长与S6态共振,因此,其共振拉曼光谱的强度模式体现了波包沿着 S6态的Franck-Condon区域势能面演化的超快动力学过程。同理,266.0、273.9和282.4 nm激发波长与S8态共振,309.1和319.9 nm激发波长与S7态共振,它们对应的共振拉曼光谱强度模式体现了在相应激发态的Franck-Condon区域的超快动力学过程。
在共振拉曼光谱中,由于高电子激发态的非共振(off-resonance)或预共振(pre-resoncne)效应28、多电子态间跃迁矩的非一致性相干28或振动-电子耦合等效应29,高电子激发态会对低激发态共振拉曼光谱的基频产生额外的增强或减弱效应。图 4可见,266.0、273.9和282.4 nm共振拉曼光谱中最强的拉曼峰为ν13和ν23,其中ν23峰强度延伸至309.1 nm共振拉曼光谱中,而ν13峰强度一直延伸至354.7 nm共振拉曼光谱中。这表明S8态对309.1和319.9 nm共振拉曼光谱的ν23峰产生明显的预共振增强效应,对309.1、319.9、341.5和354.7 nm共振拉曼光谱的ν13峰产生明显的预共振增强效应。同理,S7对341.5和354.7 nm共振拉曼光谱的ν5和ν11基频也会有一定的预共振增强效应。然而,高电子激发态会对低激发态共振拉曼光谱的倍频和组合频一般不产生预共振效应或影响不大。30因此,扣除预共振效应、排除溶剂扣减的瑕疵峰和可能的激光线影响后,从各共振拉曼光谱中能得到如下信息:

图5 24DTU在乙腈溶剂中的266.0 nm (上)、319.9 nm(中)和 341.5 nm (下)共振拉曼光谱Fig.5 266.0 nm (top), 319.9 nm (middle) and 341.5 nm(bottom) resonance Raman spectra of 24DTU in acetonitrile.
S8态短时结构动力学:266.0、273.9和282.4 nm 共振拉曼光谱可指认为 ν23、ν17、ν16、ν15、ν14、ν13、ν12、ν11、ν9、ν8、ν7、ν6和 ν5等 13 个振动基频以及它们的倍频和合频,其中,N1C6C5H11/C4C5C6H12扭转振动模 ν23、C2S8/C4S10伸缩 +C2N1C6面内弯曲振动模 ν16、C2S8/C4S10伸缩 +C4N3H9/N1C2N3面内弯曲振动模ν13、N1C2伸缩 +C2N1C6/N1C2N3/C4N3C2面内弯曲振动模 ν17、N1C6/N3C4伸缩+C6C3H11/C5C6H12面内弯曲振动模ν12和C5C6N1/C5C6H12/ C2N1C6/N1C2N3面内弯曲 +N1C6伸缩振动模ν15振动模的基频、倍频和合频,及其它们与其他振动模的合频强度占据共振拉曼光谱总强度的绝大部分。这表明,S8态短时结构动力学主要沿 N1C6C5H11/C4C5C6H12、C2S8/C4S10/N1C6/N3C4和C2N1C6/C4N3H9/N1C2N3/C5C6N1/C5C6H12/C2N1C6等多维内坐标展开。ν23归属 A′不可约表示,在S8(ππ*)态绝热动力学过程中为非拉曼活性的。因此,根据拉曼强度的振动电子耦合理论31,32和非绝热体系共振拉曼光谱研究结果33–36,强ν23(A′)振动模的出现预示 24DTU 在 S8(ππ*)态的Franck-Condon区域或附近发生了与其它势能面交叉的非绝热过程,其结构动力学涉及极快速的结构扭转运动。
S7态短时结构动力学:309.1和319.9 nm共振拉曼光谱可指认为 ν17、ν16、ν15、ν14、ν13、ν12、ν11、ν9、ν8、ν7、ν6和 ν5等 12 个振动基频以及某些较强基频的倍频和组合频。与S8态短时结构动力学不同,S7(ππ*)态短时动力学不含 A′不可约表示的振动坐标,表明在Franck-Condon区域及附近没有S(nπ*)态的强力耦合。重要的是,309.1和319.9 nm共振拉曼光谱的强度模式与266.0、273.9和282.4 nm共振拉曼光谱的明显不同,其强度模式主要由C5C6伸缩振动模 ν5、C2S8/C4S10伸缩 + C4N3H9/N1C2N3面内弯曲振动模 ν13、C2S8/C4S10伸缩 +C2N1C6面内弯曲振动模 ν16和 N2C3/C2S8/C4S10伸缩 + C6N1H7/C5C6H12面内弯曲振动模ν11等构成,说明 S7(ππ*)态短时动力学主要沿 C5C6/C2S8/C4S10/N2C3和 C4N3H9/N1C2N3/C2N1C6/C6N1H7/C5C6H12等内坐标演化。
S6态短时结构动力学:341.5和354.7 nm共振拉曼光谱可指认为 ν17、ν16、ν15、ν14、ν13、ν10、ν9、ν8、ν7、ν6和 ν5等 11 个振动基频以及它们的倍频和合频。它们的强度模式与与S7或S8态共振拉曼光谱强度模式都不同,其短时结构动力学主要沿 C5C6/N3C2/C4S10/C2S8和 C6N1H7/C5C6H12/C5C6N1/C5C6H12/C2N1C6/N1C2N3/C4N3H9/N1C2N3等内坐标演化。
5 结 论
采用共振拉曼光谱技术结合密度泛函理论计算方法研究了 24DTU 的电子激发和Franck-Condon区域结构动力学,获得如下结论:
(1) 共振拉曼光谱强度模式与去卷积获得的四个紫外吸收带的振子强度 f间存在一一对应关系。中等强度的350 nm吸收带被指认为S0→ S2跃迁(A带吸收), 338 nm (f = 0.1491)、301 nm (f =0.1795)和278 nm (f = 0.3532)强而宽的吸收带(B、C 和 D带)被分别指认为 S0→ S6、S0→ S7和 S0→S8跃迁。
(2) B带、C带和D带共振拉曼光谱可被分别指认为由11、12和13个振动基频以及它们的倍频和合频。在乙腈和甲醇中,341.5和354.7 nm共振拉曼光谱主要归属于 B带吸收或 S0→ S6跃迁;309.1和319.9 nm共振拉曼光谱主要归属于C带吸收或S0→ S7跃迁;282.4、273.9和266.0 nm共振拉曼光谱主要归属于D带吸收中或S0→ S8跃迁。
(3) S6、S7和S8态短时动力学主要沿多维内坐标展开。S8态短时结构动力学最重要的特征是在Franck-Condon区域或附件发生了S8(ππ*)/S(nπ*)势能面交叉引发的、伴随超快结构扭转运动的非绝热过程。S7和S6激发态短时动力学分别沿C5C6/C2S8/C4S10/N2C3+ C4N3H9/N1C2N3/C2N1C6/C6N1H7/C5C6H12和C5C6/N3C2/C4S10/C2S8+ C6N1H7/C5C6H12/C5C6N1/C5C6H12/C2N1C6/N1C2N3/C4N3H9/N1C2N3等内坐标演化。
(1) Crespo-Hernández, C. E.; Cohen, B.; Hare, P. M.; Kohler, B. Chem.Rev. 2004, 104 (4), 1977. doi: 10.1021/cr0206770
(2) Crespo-Hernández, C. E.; Cohen, B.; Kohler, B. Nature. 2005, 436(7054), 1141. doi: 10.1038/nature03933
(3) Middleton, C. T.; De La Harpe, K.; Su, C.; Law, Y. K.;Crespo-Hernández, C. E.; B. Kohler. Ann. Rev. Phys. Chem. 2009,60, 217. doi: 10.1146/annurev.physchem.59.032607.093719
(4) Buchvarov, I.; Wang, Q.; Raytchev, M.; Trifonov, A.; Fiebig, T.Proc. Nat. Acad. Sci. 2007, 104 (12), 4794.doi: 10.1073/pnas.0606757104
(5) Markovitsi, D.; Onidas, D.; Gustavsson, T.; Talbot, F.; Lazzarotto, E.J. Am. Chem. Soc. 2005, 127 (49), 17130. doi: 10.1021/ja054955z
(6) Kuramochi, H.; Kobayashi, T.; Suzuki, T.; Ichimura, T. J. Phys.Chem. A 2010, 114 (26), 8782-8789. doi: 10.1021/jp102067t
(7) Pollum, M.; Crespo-Hernández, C. E.; J. Phys. Chem. 2014, 140,07110. doi: 10.1063/1.4866447
(8) Pollum, M.; Jockusch, S.; Crespo-Hernández, C. E. J. Am. Chem.Soc. 2014, 136 (52), 17930. doi: 10.1021/ja510611j
(9) Taras-Goślińska, K.; Burdziński, G.; Wenska, G. J. Photochem.Photobiol. A 2014, 275, 89. doi: 10.1016/j.jphotochem.2013.11.003
(10) Harada, Y.; Suzuki, T.; Ichimura, T.; Xu, Y. J. Phys. Chem. B 2007,111 (19), 5518. doi: 10.1021/jp0678094
(11) Harada, Y.; Okabe, C.; Kobayashi, T.; Suzuki, T.; Ichimura, T.;Nishi, N.; Xu, Y. Z. J. Phys. Chem. Lett. 2009, 1 (2), 480.doi: 10.1021/jz900276x
(12) Reichardt, C.; Crespo-Hernández, C. E. J. Phys. Chem. Lett. 2010, 1(15), 2239. doi: 10.1021/jz100729w
(13) Reichardt, C.; Crespo-Hernández, C. E. Chem. Commun. 2010, 46(32), 5963. doi: 10.1039/C0CC01181A
(14) Zhang, Y.; Zhu, X.; Smith, J.; Haygood, M.; Gao, R. J. Phys. Chem.B 2011, 115 (8), 1889. doi: 10.1021/jp109590t
(15) Reichardt, C.; Guo, C.; Crespo-Hernández, C. E; J. Phys. Chem. B 2011, 115 (12), 3263-3270. doi: 10.1021/jp112018u
(16) Martínez-Fernández, L.; González, L.; Corral, I. Chem. Commun.2012, 48 (15), 2134. doi: 10.1039/C2CC15775F
(17) Cui, G.; Fang, W. J. Chem. Phys. 2013, 138 (4), 044315.doi: 10.1063/1.4776261
(18) Pollum, M,;Martínez-Fernández, L.; Crespo-Hernández, C. E.Photoinduced Phenomena in Nucleic Acid I; Springer: NY, US,1970; p 33. doi: 10.1007/128_2014_554
(19) Gobbo, J. P.; Borin, A. C.; Serrano-Andrés, L. J. Phys. Chem. B.2011, 115 (19), 6243. doi: 10.1021/jp200297z
(20) Kobayashi, T.; Kuramochi, H.; Harada, Y.; Suzuki, T.; Ichimura T.J. Phys. Chem. A 2009, 113 (44), 12088. doi: 10.1021/jp905433s
(21) Kobayashi, T.; Harada, Y.; Suzuki, T.; Ichimura, T. J. Phys. Chem. A.2008, 112 (51), 13308. doi: 10.1021/jp803096j
(22) Jiang, J.; Zhang, T. S.; Xue, J. D.; Zheng, X. M. J. Chem. Phys.2015, 143 (17), 11B605_1. doi: 10.1063/1.4935047
(23) Xie, B. B.; Wang, Q.; Guo, W. W.; Cui, G. L. Phys. Chem. Chem.Phys. 2017, 19 (11), 7689. doi: 10.1039/C7CP00478H
(24) Li, M. J.; Liu, M. X.; Zheng, X. J. Acta Phys.⁃Chim. Sin. 2013, 29(5), 903. [李明娟, 刘明霞, 郑旭明. 物理化学学报, 2013, 29 (5),903.] doi: 10.3866/PKU.WHXB201302272
(25) Frisch, M. J.; Treucks, G. W.; Schlegel, H. B.; et al. Gauss 09[M];Gaussian Inc.: Wallingford, CT, 2009.
(26) Jamróz, M. H. Spectrochim. Acta A 2013, 114, 220.doi: 10.1016/j.saa.2013.05.096
(27) Dennington, R.; Keith, T.; Millam, J. GaussView, version 5.Semichem Inc.: Shawnee Mission, KS, 2009.
(28) Galica, G. E.; Johnson, B. R.; Kinsey, J. L.; Hale, M. O. J. Phys.Chem. 1991, 95, 7994. doi: 10.1021/j100174a003
(29) Phillips, D. L.; Myers, A. B. J. Chem. Phys. 1991, 95, 226.doi: 10.1063/1.461479
(30) Biswas, N.; Umapathy, S. J. Phys. 1997, 48 (4), 937.doi: 10.1007/BF02845597
(31) Tang, J.; Albrecht, A. C. J. Chem. Phys. 1968, 49 (3), 1144.doi: 10.1063/1.1670202
(32) Tang, J, Albrecht, A. C. Raman Spectrosc. 1970, 33.doi: 10.1007/978-1-4684-3027-1_2
(33) Li, M. J.; Liu, M. X.; Zhao, Y. Y.; Pei, K. M.; Wang, H. G. J. Phys.Chem. B 2013, 117 (39), 11660. doi: 10.1021/jp403798d
(34) Fang, W. X.; Zheng, X. M.; Wang, H. G.; Zhao, Y. Y.; Guang, X.G.; Phillips, D. L.; Chen, X. B.; Fang, W. H. J. Phys. Chem. 2008,133, 134507. doi: 10.1021/jp510396y
(35) Yang, Y.; Pan, S.; Xue, J. D.; Zheng, X. D.; Phillips, D. L.; Fang, W.H. J. Raman Spectrosc. 2014, 45 (1), 105. doi: 10.1002/jrs.4420
(36) Liu, M X.; Xie, B. B.; Li, M. J.; Zhao, Y. Y.; Pei, K. M.;Wang, H. G.; Zheng, X. M. J. Raman Spectrosc. 2013, 44 (3),440. doi: 10.1002/jrs.4213
UV Absorption and Resonance Raman Spectra of 2,4-Dithiouracil
JIN Ying-Chun ZHENG Xu-Ming*
(Department of Chemistry, Zhejiang Sci-Tech University, Hangzhou 310018, P. R. China)
2,4-Dithiouracil is potentially an important photosensitizer for use in photodynamic therapy. Its photophysics when populated in the lowest excited state has been studied extensively. However, its higher light absorbing excited states and the corresponding reaction dynamics have not been investigated sufficiently. Herein, the resonance Raman spectroscopy and density functional theory were adopted to clarify the electronic transitions associated with the UV absorptions in the far-UV region and the short-time structural dynamics corresponding to the higher light absorbing excited states. The UV absorption spectrum in acetonitrile was deconvoluted into four bands∶ the moderate intense absorption band at 358 nm (f = 0.0336) (A band), the intense broad absorption bands at 338 nm (f = 0.1491), 301 nm (f = 0.1795),and 278 nm (f = 0.3532) (B, C, and D bands) respectively, on the basis of the relationship between the resonance Raman intensities and the oscillator strength f. The result was consistent with the predictions made using the time-dependent density functional theory calculations and the resonance Raman intensitypatterns. Thus, the four bands resulted from the deconvolution are assigned as the S0→ S2, S0→ S6,S0→ S7and S0→ S8transitions, respectively. The resonance Raman spectra of the corresponding B, C,and D bands are assigned and the qualitative short-time structural dynamics are obtained. The major character in the short-time structural dynamics of 2,4-dithiouracil in the S8excited state is that a non-adiabatic process via S8(ππ*)/S(nπ*) curve-crossing, accompanied with ultrafast structural distortion,takes place in or near the Franck-Condon region, while the major character in the short-time structural dynamics in the S7and S6excited state appears in the multidimensional reaction coordinates, which are mostly along the C5C6/C2S8/C4S10/N2C3bond lengths + C4N3H9/N1C2N3/C2N1C6/C6N1H7/C5C6H12bond angles for the S7excited state and the C5C6/N3C2/C4S10/C2S8bond lengths + C6N1H7/C5C6H12/C5C6N1/C5C6H12/C2N1C6/ N1C2N3/C4N3H9/N1C2N3bond angles for the S6excited state.
2,4-Dithiouracil; Excited state structural dynamics; UV absorption spectrum;Resonance Raman spectrum; Density functional calculation
April 11, 2017; Revised: May 8, 2017; Published online: May 17, 2017.
O641;O643
10.3866/PKU.WHXB201705175 www.whxb.pku.edu.cn
*Corresponding author. Email: zxm@zstu.edu.cn; Tel: +86-571-86843627.
The project was supported by the National Natural Science Foundation of China (21473163) and National Key Basic Research Program of China (973)(2013CB834607).
国家自然科学基金(21473163)和国家重点基础研究发展规划项目(973) (2013CB834607)资助
© Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
中国人民公安大学学报(自然科学版)(2022年1期)2022-07-20
新疆大学学报(自然科学版)(中英文)(2022年2期)2022-03-27
汕头大学学报(自然科学版)(2020年4期)2020-12-14
山东交通科技(2020年2期)2020-08-13
水文地质工程地质(2019年6期)2019-12-09
天然产物研究与开发(2018年5期)2018-06-13
中国有色金属学报(2018年2期)2018-03-26
电子制作(2017年20期)2017-04-26
中成药(2016年8期)2016-05-17
原子与分子物理学报(2015年3期)2015-11-24
