
氧硫化碳在230 nm光激发下的S(3P)解离通道
2017-11-01 18:11吴向坤于同坡周晓国刘世林
物理化学学报 2017年10期
吴向坤 高 治 于同坡 周晓国 刘世林
(中国科学技术大学化学物理系,合肥微尺度物质科学国家实验室,合肥 230026)
氧硫化碳在230 nm光激发下的S(3P)解离通道
吴向坤 高 治 于同坡 周晓国*刘世林
(中国科学技术大学化学物理系,合肥微尺度物质科学国家实验室,合肥 230026)
在230 nm 激光激发下,氧硫化碳(OCS)分子迅速解离生成振动基态但高转动激发的CO(X1Σg+, v = 0,J = 42–69)碎片,并通过共振增强多光子电离技术实现其离子化。通过检测处于J = 56–69转动激发态CO碎片的离子速度聚焦影像,我们获得了各转动态CO碎片的速度分布和空间角度分布,其中包含了S(1D) + CO的单重态和S(3PJ) + CO三重态解离通道的贡献。不同的转动态CO碎片对应三重态产物通道的量子产率略有不同,经加权平均我们得到230 nm附近光解OCS分子中S(3P)解离通道的量子产率为4.16%。结合高精度量化计算的OCS分子势能面和吸收截面的信息,我们获得了OCS光解的三重态解离机理,即基态OCS(X1A')分子吸收一个光子激发到弯曲的A1A'态之后,通过内转换跃迁回弯曲构型的基电子态,随后在C―S键断裂过程中与23A"(c3A")态强烈耦合并沿后者势能面绝热解离。
氧硫化碳;光解离;共振增强多光子电离;通道分支比;离子速度成像
1 引 言
氧硫化碳(OCS)是地球大气层中的重要组成部分1,是大气中组分最高的硫化合物2,3。由于它在大气中化学性质相对不活跃,因此在对流层的生存时间约为3.7年,在平流层中半衰期增加为65 ± 15年4,5。OCS的紫外光解不仅是平流层大气中二氧化硫的主要来源6,而且也是相关大气光化学反应的主要引发过程,前人为此已经在实验和理论计算方面进行了比较详细的研究7–23。OCS的电子结构同二氧化碳一样,是典型的 16个价电子结构,它们的基电子态均为线性构型,而电子激发态则可能出现线性和弯曲的两种结构,所以对氧硫化碳的光解离研究可以帮助我们深入了解此类线性三原子分子的解离机理。
OCS分子的第一吸收带从260 nm左右开始,延续至190 nm,表现为宽的类高斯谱带形状,并存在较弱的振动结构。在这一波段吸收光子后,OCS被激发解离,产生CO分子和S原子碎片,相应的解离通道研究已被广泛关注24–41。其中,主要涉及两个解离通道(如下式),分别生成单重态S(1D)和三重态S(3PJ)原子碎片,

由于通道(1)是主要解离路径,所以以往的实验和理论研究主要集中在这一单重态解离过程上。考虑到基态CO分子的平衡键长比中性OCS分子中的CO键长仅缩短了3%,所以解离生成的CO碎片更趋向于处于振动基态,这在较低解离能的实验中得到了验证26,29,42。随着解离可资用能的增加,CO碎片也有可能被振动激发,如Wei等40在214 nm光解OCS的实验中发现振动激发(v = 1)的CO碎片约占21%,而Lee等43在193 nm的光解实验中甚至观测到CO (v = 4)的解离碎片。
相对较冷的振动布居而言,解离产生CO碎片的转动激发非常明显,其转动布居显示为典型的“双钟”结构24,26,29。随着激发能的增加,两部分转动态布居的最可几分布均向更高的J值移动。此外,沿单重态通道(1)生成的CO (1Σ+g, v = 0)碎片的角分布随J量子数的变化很大24,26,27,35,38,39,43,其低J分布中各向异性参数β为较小的正值,而在高J分布区,β迅速增长接近其最大值 2。事实上,以往的量子化学和波包动力学计算24,40,41证实:OCS吸收紫外光子后,21A'和11A"两个电子激发态共同作用,生成低转动激发的CO碎片,此外21A'态还可以在弯曲构型下通过内转换至X1A",继而绝热跃迁生成高转动激发的CO碎片。总的解离机理如下式(3,4)表示:

尽管 OCS分子的第一吸收带对应的光解离已开展了大量的实验和理论研究,但是其中三重态解离通道的研究则相对较少,这主要是由于该解离通道为跃迁自旋禁阻过程,相对通道分支比较小,并且涉及更为复杂的多个电子态势能面相互作用。Sivakumar等26通过探测222–248 nm光激发下OCS解离产物CO的激光诱导荧光,第一次直接探测到三重态产物,并且利用激光诱导荧光效率估算了S(3PJ)通道分支比的上限约为2%。随后的研究27又进一步修正了三重态通道产率约为 5%,并且 222 nm光解时CO碎片的平均各向异性参数β为0.3 ±0.2。Katayanagi等28观测到223 nm处OCS光解产物 S(1D2)和 S(3P2)都具有双布居结构,低速率分布处β为1.1,而高速率处β为0.1。由于单重态和三重态通道的双布居角度分布相同,他们认为这两个通道来自相同的初始光学跃迁,其中 S(3P2)碎片通道来自单重态系间穿越(Intersystem Crossing)到三重态后的解离。Suzuki等24测量了223 nm光解OCS时产生的S(3PJ, J = 1, 2, 3)的速度分布,从中可以看出不同角动量J的S原子碎片速度分布差异很大。Sugita等31在230 nm处光解OCS,高转动态J ≥ 65产物CO的离子速度聚焦影像中明显存在三重态解离通道的贡献,由此他们认为沿三重态解离时,OCS 分子处于弯曲结构。Lipcinc和Janseen36测量了230 nm附近光解OCS的解离产物CO碎片的能态分布,给出了振动基态的 OCS分子解离生成转动态 J = 63的 CO碎片中三重态的量子产率为0.038,但遗憾的是,处于其它转动态的CO碎片中三重态量子产率未测量,从而很难准确评估总的三重态通道分支比。Brouard等38通过测量248 nm光解下产生的 S(3PJ)速度聚焦影像,给出了其速度分布和各向异性参数,其中 S(3PJ)主要来源于 21A'激发态内转换到基电子态后,再与三重态的势能面耦合解离得到。Toulson和 Murray21通过光致碎片激发谱(PHOFEX)发现三重态产物 S(3PJ)的碎片谱为在一个宽带上叠加了明显的共振结构。
相对实验而言,OCS分子激发态解离机理的理论研究不多24,40,41。最近,Schmidt等22,23应用完全活性空间自洽场(CASSCF)和多参考组态相关(MRCI)方法计算了 OCS分子最低的四个单重态和四个三重态势能面和跃迁偶极矩,展示了这些单重态和三重态之间存在着强弱不一的自旋-轨道耦合作用。这些结果不仅阐明了 OCS分子第一吸收带的共振结构来源,而且为我们进一步深入研究三重态产物通道来源提供了重要的理论参考。基于这样的设想,我们采用230 nm附近的光激发解离OCS分子,并运用高分辨的时间切片离子速度聚焦影像技术探测了处于振动基态而转动高激发态(J = 56–69)的 CO碎片,获得了相应的速度分布和角度分布。在此基础上,我们得到了各转动态CO碎片对应的三重态通道分支比,并通过与理论计算结果的比较讨论了 OCS分子在 230 nm 光激发下产生S(3P)碎片的解离机理。
2 实 验
实验设备为自行改进的三维离子速度聚焦成像系统,主要由脉冲分子束源系统、可调谐的脉冲激光系统、真空系统、离子聚焦透镜、以及质谱和影像采集和处理系统组成。实验真空腔体主要包括束源腔和电离腔两部分,分别由抽速为 2300 l∙s−1的分子泵(Agilent TV 2300,意大利)和抽速 2200 l∙s−1的分子泵(Edwards STP-A2203,日本)及前级干泵(Edwards XDS35i)实现。中间由一个直径为 1.0毫米的准直漏勺(skimmer)分开,两腔室静态真空分别为 1 × 10−5和 8 × 10−6Pa。实验中,静滞压强为3.0 × 105Pa的氧硫化碳和氩(体积比1:50)的混合气通过脉冲喷嘴(General valve, Series 9,美国)进入真空腔形成射流冷却的分子束,两腔的动态压强为4 ×10−3和 8 × 10−5Pa。
实验中,我们使用一台 Nd:YAG (Spectra Physics, LAB-190,美国)激光器的三倍频(355 nm)泵浦染料激光器(Sirah, PRSC-LG-24,德国),输出的可见光经BBO倍频产生229.82–230.00 nm的脉冲激光,单脉冲能量约为1 mJ∙pulse−1。倍频光经极化器后确定为竖直偏振的线偏光,在500 mm焦距的透镜聚焦作用下与OCS/Ar的混合气体分子束流垂直相交。OCS分子吸收光子后迅速解离,产生的CO碎片被同一束激光经共振增强多光子电离电离,离子在 25片圆电极构成的离子透镜的作用下速度聚焦投影至微通道板(Φ40 mm)及荧光屏(P47,Burle)上,发光图像由CCD相机记录。实验中,我们通过在微通道板上施加脉冲高压的方式对CO+离子进行三维时间切片的速度成像44,45,时间切片的有效脉宽约为30 ns。此外,经微通道板放大的电信号同步输出至数字示波器(Tektronics, TDS3052B,美国)平均存储,并由电脑完成飞行时间质谱(Time-of-flight Mass Spectra,TOF-MS)采集。经氧气电离解离实验测试,该成像谱仪的平动能分辨率(ΔE/E)优于 1%。
3 结果与讨论
3.1 解离产物CO的共振增强多光子电离(REM PI)谱
OCS吸收一个230 nm左右的光子后迅速解离产生CO分子及S原子碎片,其中CO分子主要布居在振动冷而转动热的内态上。如式(5)显示,产物CO可以通过(2 + 1) REMPI经中间态CO (B1Σg+)的Q支跃迁电离,从而实现转动态选择的CO+探测。
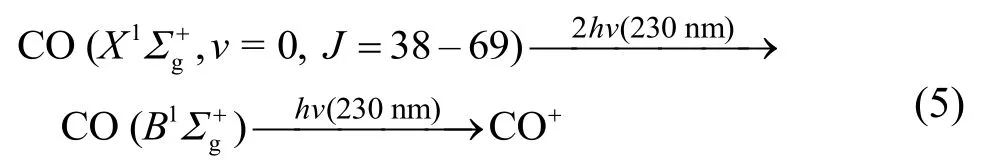
图 1(a)是光解碎片 CO (X1Σg+, v = 0, J)的(2 + 1)REMPI光谱。显然,其中存在典型的双转动态布居,中心分别处于J = 48和62处,这与前人的结果基本一致29,32。此外,由于 CO (B1Σg+, J ≥ 38)态存在预解离过程46,并且预解离速率与J值有关47,因此各转动支谱峰宽度呈现一定的加宽趋势。考虑到Q支双光子共振的跃迁强度I(J)与转动量子数J的关系如下式(6),我们可以通过比较光谱谱峰面积而得到各个转动态的相对布居比。

式中 μS、μI是跃迁因子。由于 μ2S/μ2I的真实值不超过1/1748,所以我们近似使用(2J + 1)来估算跃迁强度I (J)时,在J = 38–70的范围内误差不超过0.3%。应用图1(a)的转动态谱峰强度除以跃迁强度I (J)即可得到转动布居比,在对总的光谱强度做归一化处理后,图1(b)即为得到的光解碎片CO (X1Σ+g, v = 0,J)的转动布居PJ。
3.2 解离碎片CO的共振增强多光子电离质谱
选择激发波长为 229.956,229.868和 229.849 nm,探测解离碎片CO的共振增强多光子电离质谱如图2所示。由图1可知,这些波长正好与OCS解离生成CO碎片的双光子共振跃迁B1Σg+(v = 0, J)←X1Σg+(v = 0, J)的Q支共振,J分别为49、63和68。图 2的飞行时间质谱中存在唯一的碎片离子CO+,并且在较低的转动态(J = 49)时质谱峰形类似于矩形,而当J增大至68时谱峰明显分为两个部分,即宽的包络平台上叠加了尖锐的谱峰。由此,我们可以推测,OCS光解产生高转动激发CO碎片的过程存在多个解离通道,产生了不同的平动能分布。

图1 (a)光解碎片CO v = 0, J)的(2 + 1)REMPI光谱;(b)光解碎片 CO( v = 0, J)的转动态相对布居PJ (J = 38–70)Fig.1 (a) (2 + 1) REMPI of CO(v = 0, J) fragment; (b) relative population, PJ (J = 38–70), of CO v = 0, J)fragment.
如图2所示,由于OCS解离时富余能的释放,CO (X1Σg+)碎片离子谱峰的宽度显著加宽。在当前的引出电场强度(48 V∙cm−1)下,CO+谱峰的全宽分别为 250 (J = 49)、282 (J = 63)和 345 ns (J = 68)。考虑到我们的脉冲质量门仅~30 ns,远小于CO+谱峰宽度,因此时间切片的离子聚焦速度图像可以直接反映CO碎片的三维速率分布和空间角度分布,无需做反阿贝尔变换。由于我们研究的重点是产生S(3P)的解离通道,因此主要测量和关注了高转动态(J ≥55)的CO (X1Σg+, v = 0, J)碎片的能量布居和空间分布。

图2 OCS分子在229.956,229.868,229.848 nm处光解离-电离的飞行时间质谱Fig.2 Time-of-flight mass spectra of dissociative photoionization of OCS at 229.956, 229.868 and 229.848 eV.
图3是解离碎片高转动态CO (X1Σ+, v = 0, J =55–69)的三维速度切片影像。各影像图中,激光的电场矢量ε为竖直方向,平行于探测器平面,每张图像均累积了至少 10万个点。大部分影像可以清楚地分为两部分,一部分是处于中心的多个同心圆环,另一部分则是亮度较弱的外部大圆环,并且各圆环都表现出较为明显的各向异性。随着J值的增加,内环和外环的半径均在逐渐减小,并且外环亮度越来越明显。这样,三维速度切片影像清楚地暗示着OCS分子在230 nm附近的光解离至少存在两种解离通道,并且通道分支比显著依赖于转动的激发。

图3 OCS 光解产生的解离碎片CO (X1Σg+, v = 0, J)的速度聚焦时间切片影像Fig.3 Time-sliced velocity map images of CO (X1Σg+, v =0, J) fragment dissociated from OCS.
针对图3的离子影像,我们可以通过对相同半径的图像强度积分来获得处于各转动态的CO碎片速率分布。经过离子影像的能量校准,我们获得的典型CO碎片的速率分布如图4所示。
解离过程中,碎片的能量守恒关系如下(7)式所示,

其中,hν是光子能量,D0是解离限,解离通道(1)为4.279 eV,解离通道(2)则为3.120 eV,Evib(OCS)和Erot(OCS)则分别是OCS分子的初始振动能和转动能,Eava是解离的可资用能,ET(CO)和 ET(S)是CO和S原子的平动能,Evib(CO)和Erot(CO)分别是CO碎片的振动和转动能量。实验中采用的是超声分子束,束流平动温度约为10 K,因此OCS的初始转动温度也近似被认为是10 K,这样Erot(OCS)可以忽略不计。应用上述的能量守恒和动量守恒(mCO·vCO= mS·vS)关系,我们可以在测量的CO碎片速率基础上得到解离过程中的总平动能释放状况。参考前人的结论31,32,36,图3中的内环被归属为来自单重态解离通道(1)。各内环对应的总能量间隔为0.066 eV,这与OCS母体分子的弯曲振动频率(v2=0.065 eV)极为接近31,32,36,并且各图中的不同内环对应OCS解离的解离限D0一致,因此这些同心圆环可以被认为是分别来自母体OCS分子的初始v2弯曲振动激发,由内而外分别对应振动量子数 v2为0,1和2,这与前人的主要结论保持一致31,32,36。类似地,图像中外侧大圆环对应较大的碎片平动能,依据能量守恒,该过程对应更低的解离限,我们将其归属为沿通道(2)解离产物的贡献31。因为速度聚焦图像的分辨和图像的半径存在非线性依赖关系,半径越大,分辨相对越差,因此当前图像中外环中不能清楚地分辨出OCS分子的初始v2弯曲振动(但依然隐约可见,如图4)。
此外,随着转动量子数J的增加,通道(1)的同心圆环中,OCS基振动态(v2= 0)的贡献越来越少,尤其是在J = 69时,基振动态(v2= 0)的贡献基本消失,这意味着 OCS的弯曲振动激发更有利于解离产生高转动的CO碎片。
根据图4所示的平动能分布,我们可以得到CO碎片处于J = 55–69之间的转动态时,沿三重态产物通道解离时平动能的释放占可资用能的比例约为65.69%–50.98%。如果这一解离为快速的直接解离过程,那么我们可以用经典的反冲模型49来解释,即采用式(8)来估算平均平动能占可资用能的比例fT。

这样,反冲模型预测的 OCS分子快速解离得到CO碎片的过程fT为58.5%,这与当前的实验结果非常相似,这侧面说明了该三重态产物通道(2)为快速直接解离机制。
3.4 三重态量子产率及角分布
通过积分图4中三重态通道的速率分布(即图3的外圆环)强度并归一化,我们可以得到OCS分子解离产生的 CO各转动态对应的三重态通道分支比。由于脉冲场时间切片时,相同脉冲宽度下内圆环的切片比例较外环大,这样直接利用时间切片影像得到的信号强度来计算通道分支比的结果较真实值偏小。为此,我们通过电场拟合,充分考虑了外环和内环的半径及切片的宽度的影响,对三重态通道分支比的实验结果进行了修正,修正后的各转动态 CO对应的单重态和三重态产物通道分支比TPJ如图 5所示。显然,该分支比严重依赖于 CO碎片的转动激发程度,因此Lipciuc和Janseen36只用J = 63的通道分支比代表OCS光解离得到三重态产物分支比是不合适的。
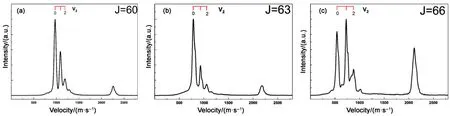
图4 OCS光解产生的解离碎片CO (X1Σg+, v = 0, J)的速度分布Fig.4 Speed distributions of the CO fragment dissociation from OCS.(a) J = 60; (b) J = 63; (c) J = 66.
总体上,三重态通道分支比TPJ在J ≥ 63之后迅速增加,这意味着OCS分子的高弯曲激发能够更有效地促进三重态的产物生成。为了更为准确地获得230 nm附近OCS分子光解三重态通道的分支比,我们采用了加权平均的方法,利用解离得到各转动态CO碎片的三重态比例TPJ乘以该转动态的相对布居PJ(图1(b)),然后对J值求和,即

由此,我们得到的三重态产物通道分支比为4.16%,这与Nan等27在222 nm处光解估计的5%比较接近。
通过积分速度聚焦影像(图 3)中各角度下一定范围的速度分布,我们可以得到解离碎片CO的角度分布I(θ),对应的各向异性参数β用下式50拟合得到,

图5 来自单重态和三重态光解通道的碎片CO (X1Σ+g, v = 0, J = 55−69)相对比例 TPJFig.5 Branching ratio of the singlet and triplet dissociation channels TPJ of CO (X1Σ+g, v = 0, J = 55−69)fragment.
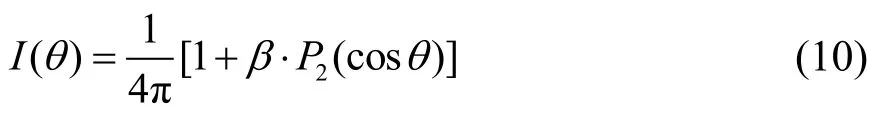
其中θ是碎片反冲速度和解离光的电场矢量ε之间的夹角,P2(cosθ)是二阶勒让德函数。拟合得到的各解离通道β值整理在表1中,其中我们也列出部分以前的实验结果作为对比。显然,各态的结果与以往实验结果基本一致,并且三重态解离通道的 β值随CO碎片的转动量子数J的增大而减小,且始终为正值,这意味着三重态解离通道主要来自于OCS的平行跃迁过程。
3.5 230 nm附近OCS分子的解离机理
Schmidt等22,23计算的OCS最低的四个单重态和三重态的势能面如图6所示。由于OCS分子基电子态是单重态,因此自旋允许跃迁的单重态解离通道,CO(X1Σ+) + S(1D),相对分支比较大,而自旋禁阻的三重态解离通道 CO(X1Σ+) + S(3PJ)几率很小,通道分支比低。在230 nm光子能量附近,OCS分子可以被激发到21A′(A1A′)和 11A′(B1A′)两个单重排斥态直接解离。Schmidt等22,23计算得到A1A′ ←X1A′跃迁截面为 1.73 × 10−19cm2,远大于 B1A′ ←X1A′的跃迁截面(1.4 × 10−21cm2),所以 A1A′态在单重态解离通道中占主导地位,这与Suzuki等24的量化计算结论一致。波包模拟计算更是确认了单重态解离中高转动激发的CO碎片仅来自于21A'的贡献。此外,由于这两个单重态都是弯曲的,所以跃迁中OCS分子几何结构发生了从线性到弯曲的显著变化。

表1 OCS光解碎片CO (X1Σ+g, v = 0, J = 55–69)的各向异性参数Table 1 Anisotropy parameters of CO (X1Σ+g, v = 0, J = 55–69)fragment dissociated from OCS.
在230 nm光激发下,由于自旋-轨道耦合的作用,跃迁禁阻的三重态 13A′(a)、13A′(b)、23A′(c)和23A'(d)态都有可能被激发。如图 6所示,23A′(d)是典型的束缚电子态,与实验观测的快速解离性质不符,所以实验中测量的S(3P)三重态产物只能来自于13A′(a)、13A′(b)或 23A′(c)电子态。依据 Schmidt等的计算结果,在考虑自旋-轨道耦合作用后,这三个态的吸收截面分别为 0.000292,0.00906和 0.129(10−19cm2),因此 23A′(c)电子态是导致 OCS 分子第一吸收带振动结构的主要原因。类似的,我们推测实验中所观测到的三重态产物主要来自于 23A′(c)电子态的解离。
然而,Schmidt等计算显示 23A′(c)电子态沿R(C―S)键长变化方向上有大约 6 eV 的势垒(如图6)。考虑到230 nm光子的激发能仅为5.4 eV, 直接吸收一个光子无法越过该势垒解离。不仅如此,由基态至c态的跃迁为A′ → A′,是典型的垂直跃迁,各向异性参数β应该是负值,这与我们的实验结果正好相反。因此,当前实验中的三重态解离产物不会来自 X1A′ → c3A′的 Franck-Condon 跃迁。
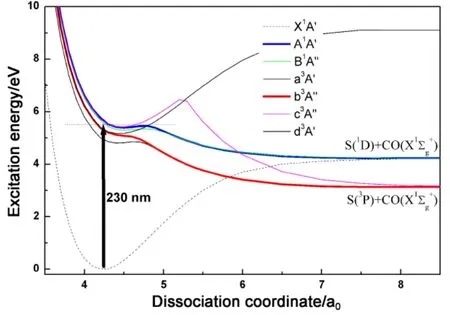
图6 OCS分子沿解离坐标R(C―S)键长变化的势能曲线22Fig.6 Potential energy curves of OCS along with the C―S bond22.
如表1所示,单重态的解离通道具有较大的各向异性参数(正值),因此主要来自于 A1A′ ← X1A′的跃迁,这与吸收截面的大小顺序相符。Suzuki等24认为高转动激发的CO碎片对应的单重态解离通道来自于A1A′在弯曲构型下内转换到基态X1A′后绝热解离,这一结论得到了Wei等40和Mcbane等41的计算证实。考虑到在我们实验中,三重态解离通道的各向异性参数远大于0且随CO碎片的J值增大而减小,这与单重态解离通道非常相似,因此我们有理由相信这些三重态解离通道产物与相应的单重态解离过程具有相同的初始光学跃迁,即由X1A′激发至弯曲的A1A′态后,在弯曲构型下通过内转换跃迁回 X1A′的高振动态,在较大的 C-S距离处(例如,≥ 5.5 a0)与 23A′(c)态耦合并沿后者势能面绝热解离,

4 结 论
我们应用230 nm光激发OCS分子产生了CO解离碎片,并利用离子速度聚焦影像技术测量了这些处于振动基态但高转动激发态(J = 55–69)的 CO分子能量分布和空间分布。各转动态CO分子的影像中存在明显的两部分,较小半径的若干同心圆环和外部较大半径的圆环。前者被归属为单重态解离通道的贡献,即CO(X1Σ+) + S(1D),而后者则对应三重态解离通道,CO(X1Σ+) + S(3P)。
通过对各转动态CO的离子速度聚焦影像的处理,我们得到各转动态CO碎片对应的平动能分布和空间分布,并且可以用经典的反冲模型来近似模拟。有意思的是,各转动态CO对应的单重态和三重态产物通道分支比严重依赖于CO碎片的转动激发程度,因此Lipciuc和Janseen36采用J = 63的通道分支比代表总产物分支比是不合适的。通过对时间切片宽度影响的修正,我们得到了各转动态 CO碎片对应的三重态与单重态解离通道的量子产率,并计算得到总的通道分支比为4.16%。
此外,通过拟合各转动态CO的角度分布,我们还获得了单重态和三重态通道的各向异性参数。结果显示三重态解离通道与单重态类似,也来自于平行跃迁,并且两者非常接近的变化趋势意味着单重态和三重态解离通道具有相同的初始光学跃迁。基于Schmidt等22,23计算的势能面和吸收截面,我们认为在230 nm附近激发时,OCS分子的三重态解离过程来自于A1A′ ← X1A′初始激发,随后在弯曲构型下通过内转换跃迁至X1A′的高振动态,继而与23A′(c)态耦合解离导致。
(1) Hanst, P. L.; Spiller, L. L.; Watts, D. M.; Spence, J. W.; Miller, M. F.J. Air Pollut. Control Assoc. 1975, 25, 1220.doi: 10.1080/00022470.1975.10470199
(2) Montzka, S.; Aydin, M.; Battle, M.; Butler, J.; Saltzman, E.; Hall, B.;Clarke, A.; Mondeel, D.; Elkins, J. J. Geophys. Res. Atmos. 2004,109, D22. doi: 10.1029/2004JD004686
(3) Cagnioncle, A. -M.; Parmentier, E. M.; Elkins-Tanton, L. T.J. Geophys. Res. Solid Earth 2007, 112, B9.doi: 10.1029/2007JB004934
(4) Andreae, M. O.; Crutzen, P. J. Science 1997, 276, 1052.doi: 10.1126/science.276.5315.1052
(5) Krysztofiak, G.; Té, Y. V.; Catoire, V.; Berthet, G.; Toon, G. C.;Jégou, F.; Jeseck, P.; Robert, C. Atmosphere-Ocean 2015, 53, 89.doi: 10.1080/07055900.2013.876609
(6) Brühl, C.; Lelieveld, J.; Crutzen, P. J.; Tost, H. Atmos. Chem. Phys.2012, 12, 1239. doi: 10.5194/acp-12-1239-2012
(7) Forbes, G. S.; Cline, J. E. J. Am. Chem. Soc. 1939, 61, 151.doi: 10.1021/ja01870a049
(8) Sidhu, K.; Csizmadia, I.; Strausz, O.; Gunning, H. J. Am. Chem. Soc.1966, 88, 2412. doi: 10.1021/ja00963a009
(9) Breckenridge, W.; Taube, H. J. Chem. Phys. 1970, 52, 1713.doi: 10.1063/1.1673209
(10) Ferro, B.; Reuben, B.. Trans. Faraday Soc. 1971, 67, 2847.
(11) Rudolph, R. N.; Inn, E. C. J. Geophys. Res. Oceans 1981, 86, 9891.doi: 10.1029/JC086iC10p09891
(12) Molina, L.; Lamb, J.; Molina, M. Geophys. Res. Lett. 1981, 8, 1008.doi: 10.1029/GL008i009p01008
(13) Zhao, Z.; Stickel, R.; Wine, P. Geophys. Res. Lett. 1995, 22, 615.doi: 10.1029/95GL00170
(14) Wu, C. R.; Chen, F.; Judge, D. J. Quant. Spectrosc. Radiat. Transf.1999, 61, 265.
(15) Colussi, A. J.; Leung, F.-Y.; Hoffmann, M. R. Environ. Chem. 2004,1, 44. doi: 10.1071/EN04010
(16) Danielache, S. O.; Nanbu, S.; Eskebjerg, C.; Johnson, M. S.; Yoshida,N. J. Chem. Phys. 2009, 131, 024307. doi: 10.1063/1.3156314
(17) Hattori, S.; Danielache, S.; Johnson, M. S.; Schmidt, J. A.;Kjaergaard, H. G.; Toyoda, S.; Ueno, Y.; Yoshida, N. Atmos. Chem.Phys. 2011, 11, 10293. doi: 10.5194/acp-11-10293-2011
(18) Sunanda, K.; Rajasekhar, B.; Saraswathy, P.; Jagatap, B. J. Quant.Spectrosc. Radiat. Transf. 2012, 113, 58.doi: 10.1016/j.jqsrt.2011.09.009
(19) Limão-Vieira, P.; Da Silva, F. F.; Almeida, D.; Hoshino, M.; Tanaka,H.; Mogi, D.; Tanioka, T.; Mason, N.; Hoffmann, S.; Hubin-Franskin,M. -J. J. Chem. Phys. 2015, 142, 064303. doi: 10.1063/1.4907200
(20) Grosch, H.; Fateev, A.; Clausen, S. J. Quant. Spectrosc. Radiat.Transf. 2015, 154, 28. doi: 10.1016/j.jqsrt.2014.11.020
(21) Toulson, B. W.; Murray, C. J. Phys. Chem. A 2016, 120, 6745.doi: 10.1021/acs.jpca.6b06060
(22) Schmidt, J. A.; Johnson, M. S.; McBane, G. C.; Schinke, R. J. Chem.Phys. 2012, 136, 131101. doi: 10.1063/1.3701699
(23) Schmidt, J. A.; Johnson, M. S.; McBane, G. C.; Schinke, R. J. Chem.Phys. 2012, 137, 054313. doi: 10.1063/1.4739756
(24) Suzuki, T.; Katayanagi, H.; Nanbu, S.; Aoyagi, M. J. Chem. Phys.1998, 109, 5778. doi: 10.1063/1.477200
(25) Sivakumar, N.; Burak, I.; Cheung, W.; Houston, P.; Hepburn, J.J. Phys. Chem. 1985, 89, 3609. doi: 10.1021/j100263a008
(26) Sivakumar, N.; Hall, G.; Houston, P.; Hepburn, J.; Burak, I. J. Chem.Phys. 1988, 88, 3692. doi: 10.1063/1.453869
(27) Nan, G.; Burak, I.; Houston, P. Chem. Phys. Lett. 1993, 209, 383.doi: 10.1016/0009-2614(93)80035-N
(28) Katayanagi, H.; Mo, Y.; Suzuki, T. Chem. Phys. Lett. 1995, 247, 571.doi: 10.1016/0009-2614(95)01253-2
(29) Sato, Y.; Matsumi, Y.; Kawasaki, M.; Tsukiyama, K.; Bersohn, R.J. Phys. Chem. 1995, 99, 16307. doi: 10.1021/j100044a017
(30) Mo, Y.; Katayanagi, H.; Heaven, M. C.; Suzuki, T. Phys. Rev. Lett.1996, 77, 830. doi: 10.1103/PhysRevLett.77.830
(31) Sugita, A.; Mashino, M.; Kawasaki, M.; Matsumi, Y.; Bersohn, R.;Trott-Kriegeskorte, G.; Karl-Heinz, G. J. Chem. Phys. 2000, 112,7095. doi: 10.1063/1.481324
(32) Katayanagi, H.; Suzuki, T. Chem. Phys. Lett. 2002, 360, 104.doi: 10.1016/S0009-2614(02)00788-1
(33) Van den Brom, A. J.; Rakitzis, T. P.; van Heyst, J.; Kitsopoulos, T. N.;Jezowski, S. R.; Janssen, M. H. J. Chem. Phys. 2002, 117, 4255.doi: 10.1063/1.1496464
(34) Rakitzis, T. P.; van den Brom, A. J.; Janssen, M. H. Science 2004,303, 1852. doi: 10.1126/science.1094186
(35) Kim, M. H.; Li, W.; Lee, S. K.; Suits, A. G. Can. J. Chem. 2004, 82,880. doi: 10.1139/V04-072
(36) Lipciuc, M. L.; Janssen, M. H. Phys. Chem. Chem. Phys. 2006, 8,3007. doi: 10.1039/b605108a
(37) Brouard, M.; Green, A.; Quadrini, F.; Vallance, C. J. Chem. Phys.2007, 127, 084304. doi: 10.1063/1.2757618
(38) Brouard, M.; Quadrini, F.; Vallance, C. J. Chem. Phys. 2007, 127,084305. doi: 10.1063/1.2757619
(39) Lipciuc, M. L.; Rakitzis, T. P.; Meerts, W. L.; Groenenboom, G. C.;Janssen, M. H. M. Phys. Chem. Chem. Phys. 2011, 13, 8549.doi: 10.1039/c0cp02671a
(40) Wei, W.; Wallace, C. J.; McBane, G. C.; North, S. W. J. Chem. Phys.2016, 145, 024310. doi: 10.1063/1.4955189
(41) McBane, G. C.; Schmidt, J. A.; Johnson, M. S.; Schinke, R. J. Chem.Phys. 2013, 138, 094314. doi: 10.1063/1.4793275
(42) Rijs, A. M.; Backus, E. H.; de Lange, C. A.; Janssen, M. H.;Westwood, N. P.; Wang, K.; McKoy, V. J. Chem. Phys. 2002, 116,2776. doi: 10.1063/1.1434993
(43) Lee, S. K.; Silva, R.; Thamanna, S.; Vasyutinskii, O. S.; Suits, A. G.J. Chem. Phys. 2006, 125, 144318. doi: 10.1063/1.2357948
(44) Gebhardt, C. R.; Rakitzis, T. P.; Samartzis, P. C.; Ladopoulos, V.;Kitsopoulos, T. N. Rev. Sci. Instrum. 2001, 72, 3848.doi: 10.1063/1.1403010
(45) Tang, X.; Zhou, X.; Niu, M.; Liu, S.; Sun, J.; Shan, X.; Liu, F.; Sheng,L. Rev. Sci. Instrum. 2009, 80, 113101. doi: 10.1063/1.3250872
(46) Tjossem, P. J.; Smyth, K. C. J. Chem. Phys. 1989, 91, 2041.doi: 10.1063/1.457064
(47) Rottke, H.; Zacharias, H. Opt. Commun. 1985, 55, 87.doi: 10.1016/0030-4018(85)90306-2
(48) Bray, R.; Hochstrasser, R. -M. Mol. Phys. 1976, 31, 1199.doi: 10.1080/00268977600100931
(49) Holdy, K. E.; Klotz, L. C.; Wilson, K. R. J. Chem. Phys. 1970, 52,4588. doi: 10.1063/1.1673690
(50) Zare, R. N. Mol. Photochem 1972, 4, 1.
S(3P) Fragmentation Channel of Carbonyl Sulfide at 230 nm
WU Xiang-Kun GAO Zhi YU Tong-Po ZHOU Xiao-Guo*LIU Shi-Lin
(Hefei National Laboratory for Physical Sciences at the Microscale, Department of Chemical Physics,University of Science and Technology of China, Hefei 230026, P. R. China)
Carbonyl sulfide (OCS) was photoexcited at 230 nm so that it dissociated into a vibrationally cold but rotationally hot CO (X1Σg+,, v = 0, J = 42–69) fragment, which was eventually subjected to resonance enhanced multiphoton ionization. The kinetic energy release distribution and angular distribution of the CO fragment were obtained by detecting the time-sliced velocity map images of CO+in various rotational states (J = 55–69), wherein both the singlet dissociation channel of S(1D) + CO and the triplet pathway of S(3PJ) + CO were involved. For the triplet fragment channel, the total quantum yield of OCS dissociation at 230 nm was estimated to be 4.16%, based on the measured branching ratioin every rotational state. High-level quantum chemical calculations on the potential energy surface and the absorption cross section of OCS revealed the dissociation mechanism along the triplet channel of OCS,with photolysis at 230 nm. The ground state OCS (X1A') is photoexcited to the bent A1A' state at 230 nm,which then decays back to X1A' in a bent structure via internal conversion and subsequently couples to the 23A"(c3A") state by spin-orbit coupling, followed by direct dissociation along its potential energy surface.
Carbonyl sulfide (OCS); Photodissociation; Resonance enhanced multiphoton ionization; Branching ratio; Ion velocity imaging
April 22, 2017; Revised: May 10, 2017; Published online: May 18, 2017.
O643
10.3866/PKU.WHXB201705183 www.whxb.pku.edu.cn
*Corresponding author. Email: xzhou@ustc.edu.cn; Tel/Fax: +86-551-63600031.
The project was supported by the National Natural Science Foundation of China (21373194 and 21573210), National Key Research and Development Program(2016YFF0200502), National Key Basic Research Program of China (973) (2013CB834602) and the Ministry of Science and Technology of China
(2012YQ220113).
国家自然科学基金(21373194, 21573210), 国家重点研发计划(2016YFF0200502), 国家重点基础研究发展规划项目(973) (2013CB834602)和国家重大科学仪器专项(2012YQ220113)资助
© Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
黑龙江大学自然科学学报(2022年1期)2022-03-29
数学物理学报(2021年4期)2021-08-30
粉末冶金技术(2021年3期)2021-07-28
天然产物研究与开发(2019年10期)2019-11-05
学生天地(2019年28期)2019-08-25
电子制作(2019年12期)2019-07-16
中国民族医药杂志(2016年2期)2016-05-14
中国医疗美容(2015年2期)2015-07-19
原子与分子物理学报(2014年4期)2014-02-28
疯狂英语·口语版(2013年1期)2013-01-31
