
基于CRISPR/Cas9系统的人成神经瘤SK-N-SH细胞CAPNS1基因的靶向敲除
2018-09-28 09:18朱家佳龙鼎新
生命科学研究 2018年4期
朱家佳,龙鼎新
(南华大学公共卫生学院,中国湖南衡阳421001)
钙蛋白酶(calpain)是一组高度保守的特异性Ca2+依赖性细胞内中性蛋白酶,其活性主要与细胞内Ca2+浓度有关[1,2]。Calpain在机体的生理过程中发挥着重要作用,其不仅与细胞内蛋白质的水解有关,还参与细胞自噬、细胞周期调控与凋亡、细胞骨架重构、葡萄糖转运、细胞信号转导等正常的生理过程[3,4]。自Guroff[5]在大鼠的脑组织中首次发现一种可溶性Ca2+依赖的中性蛋白酶以来,研究者们越来越关注这种蛋白酶在体内的作用。目前,研究发现在哺乳动物中calpain存在15种亚型,其中,calpain1(μ-calpain)和 calpain2(m-calpain)在体内广泛表达,且研究也较为广泛[6]。Calpain1和calpain2主要由80 kD的大亚基和30 kD的小亚基(calpain small subunit 1,CAPNS1)组成,CAPNS1是calpain1和calpain2活性所必需的,而且也是细胞迁移、凋亡和存活所必需的[7]。
基因编辑技术是指在基因组水平精确地进行基因编辑,其是通过精确识别靶细胞DNA片段中靶点的核苷酸序列,利用核酸内切酶对DNA靶点序列进行切割,从而完成对靶细胞DNA目的基因片段的敲除、加入等过程。继锌指核酸酶(zincfinger nuclease,ZFN)和转录激活因子样效应物核酸酶(transcription activator-like effector nuclease,TALEN)基因编辑技术之后,第三代基因编辑技术CRISPR/Cas技术是目前运用最为广泛的基因技术。相比ZFN和TALEN,CRISPR/Cas技术具有成本低、可同时进行多位点编辑、效率高等优势[8]。CRISPR/Cas系统是细菌或古细菌中的一种适应性免疫系统,用以抵御外来质粒或噬菌体的入侵[9]。根据Cas基因序列及蛋白质结构,CRISPR/Cas系统可分为3种类型,其中Ⅰ型和Ⅲ型CRISPR/Cas系统需要多个Cas蛋白形成复合体切割DNA双链;而Ⅱ型CRISPR/Cas系统即CRISPR/Cas9系统只需要一个Cas9蛋白核酸酶即可对DNA双链进行切割,因此其最为简单并被广泛地应用于生命科学研究。该系统主要包含非特异性Cas9核酸酶和sgRNA两部分,其中sgRNA具引导和检测作用;Cas9是一种RNA依赖的DNA内切核酸酶,其利用单一向导RNA(sgRNA)使双链DNA断裂,起到剪刀的作用[10]。
本课题组在前期研究中发现calpain参与了细胞自噬的调节过程(结果尚未发表)。为进一步研究其机制,本文利用CRISPR/Cas9技术进行CAPNS1基因敲除,为后期研究calpain在自噬中的作用提供一定的实验基础。
1 材料和方法
1.1 材料
pGK1.1载体(江苏吉锐生物有限公司)、Bbs I内切酶和buffer(NEB公司,美国)、T4 DNA ligase和 buffer(Thermo Fisher Scientific 公司,美国)、质粒抽提试剂盒(Axygen公司,美国)、Top10感受态细胞[生工生物工程(上海)股份有限公司]、SK-NSH细胞(中国科学院上海生命科学研究院细胞库)、MEM 培养基(Sigma公司,美国)、胎牛血清(Biological Industries公司,以色列)、Genloci基因抽提试剂盒和CruiserTM酶(江苏吉锐生物有限公司)、BCA试剂盒(上海碧云天生物技术有限公司)、mouse anti-calpain1(Sigma 公司,美国)、rabbit anti-calpain2和rabbit anti-CAPNS1(Abcam公司,英国)。
1.2 方法
利用 NCBI(http://www.ncbi.nlm.nih.gov/)数据库获得CAPNS1基因在染色体的信息以及所有转录本公共的CDS区,同时找出公共CDS区所处的外显子进行靶位点设计。
1.2.1 CRISPR/Cas9敲除靶位点设计和寡核苷酸链合成
根据CRISPR/Cas9敲除靶位点即gRNA设计原则,通过在线软件(http://crispr.mit.edu/)设计靶序列,然后根据分值大小筛选出CAPNS1外显子区域3′端有PAM元件NGG(N为任意的核苷酸)的20 bp连续的碱基序列。PAM序列区是CRISPR/Cas9系统发挥作用的基本条件,如果靶序列没有PAM序列,Cas9蛋白将不会对靶位点进行切割[11]。根据oligo DNA进行引物合成,引物合成需在靶序列头部添加额外的碱基,其中正向引物添加cacc,反向引物添加aaac。靶序列的第一个碱基必须是G,若不是,可自行在靶序列前加一个G。
1.2.2 pGK1.1-sgRNA敲除载体构建
将合成后的两条单链oligoDNA稀释成10μmol/L,退火形成dsDNA,再与线性化后的pGK1.1载体连接。退火反应体系如下:正负链oligo各10 μL,10× annealing buffer 4 μL,ddH2O 16 μL,总体积40 μL。将以上体系瞬时离心,置于PCR仪中95℃孵育3 min,然后自然冷却至温度下降到40℃以下,获得充分杂交的DNA。取1 μL杂交后的ds-DNA进行T4 DNA ligase连接反应,反应体系如下:pGK1.1 线性质粒 1 μL,杂交后的 dsDNA 1 μL,T4 DNA ligase 0.5 μL,10× T4 DNA ligase buffer 1 μL,ddH2O 6.5 μL,总体积 10 μL。将以上体系瞬时离心后,放于PCR仪中16℃孵育30 min进行连接反应。然后转化至Top10感受态细胞中,均匀涂布于含Kan抗性的筛选平板上,倒置培养过夜。使用上游引物VSP primer与下游负链oligo引物进行菌落PCR筛选,阳性克隆PCR正确大小应为100 bp。对筛选到的阳性克隆进行质粒提取并送往南京一道生物科技有限公司进一步测序验证。
1.2.3 电转染SK-N-SH细胞及单克隆的制备和生长
SK-N-SH细胞培养于含10%胎牛血清MEM培养基、5%CO2、37℃恒温箱条件下。转染前,取状态良好的对数生长期SK-N-SH细胞悬液进行台盼蓝计数,确定细胞数及细胞活力(细胞活力>95%)。取5×106个细胞于15 mL离心管中,以转速1 000 r/min离心4 min,弃上清。将细胞沉淀悬浮于210 μL DPBS中,转移至1.5 mL EP管中,加入CAPNS1基因敲除质粒5~8 μg(质粒浓度要求1 g/L以上),轻轻混匀,进行共转染。24 h后,加入含最佳嘌呤霉素筛选浓度(1 μg/mL)的完全培养基进行药筛,2~5 d后,利用有限稀释法使最终96孔板中每孔细胞数为1个。1周后观察单克隆生长情况,约两周后将长起来的单克隆转移至48孔中扩大培养;当细胞长满48孔1/2时,即可取出一部分(102~104),使用Genloci TNA抽提试剂盒(cat.no.GP0155,GP0156)提取细胞基因组。
1.2.4 筛选基因敲除阳性克隆
PCR杂交获得杂交DNA,其反应条件如下:野生型DNA和突变型DNA各25 ng,2.5 μL 10×G-Taq buffer,1.5 μL Mg2+buffer,0.5 μL dNTP(10 mmol/L),上游引物(AAGCCATGGAGACACTATGC)和下游引物(TGGTCCATAGAGGTCATAGG)各1 μL,0.25 μL G-Taq DNA聚合酶。PCR仪中95℃预变性1.5 min,变性10 s,55~62℃退火10 s,72℃延伸20 s,彻底延伸5 min,此循环进行35次。自然冷却至40℃以下,获得杂交DNA。取2 μL PCR产物、2 μL CruiserTMbuffer、1 μL CruiserTM酶,双蒸水补充至10 μL。45℃反应20 min后立即向上述 10 μL 反应体系内加入 2 μL 6× stop buffer,随后进行琼脂糖凝胶电泳检测或置于-20℃保存。将CrusierTM酶切初步筛选出来的阳性克隆送往南京一道生物科技有限公司进行测序验证,进一步确认阳性克隆。对于两个等位基因突变情况不一样的阳性克隆,重新做TA克隆后送测序,跟野生型比对,确定每个等位基因的突变情况。
1.2.5 Western-blot检测
提取野生型SK-N-SH细胞(CAPNS1+/+)和突变型SK-N-SH细胞(CAPNS1-/-)的总蛋白质,用BCA试剂盒检测蛋白质浓度。将蛋白质样品进行SDS-PAGE电泳分离,转至PVDF膜上,5%脱脂牛奶封闭2 h,然后分别加入mouse anti-calpain1(1∶1 000)、rabbit anti-calpain2(1∶1 000)、rabbit anti-CAPNS1(1∶1 000)、rabbit anti-actin(1∶1 000),4℃孵育过夜。吸走一抗,TBST洗3遍,加入二抗(1∶3 000)37℃孵育1 h,TBST洗5遍。最后用凝胶成像系统进行显影,检测蛋白质表达情况。
2 结果
2.1 CAPNS1基因信息和靶位点设计与合成
从NCBI中获得人CAPNS1基因(ID:826)位于19号染色体(NC_000019.10),具有4套不同的转录本,序列号分别为:NM_001003962.2、NM_001302632.1、NM_001302633.1、NM_001749.3。选取CAPNS1基因所有转录本公共的CDS区域中的外显子进行靶位点设计,所获得的3个sgRNA靶位点序列信息及相对应的靶标寡核苷酸序列见表1。
2.2 pGK1.1载体图谱及酶切鉴定
pGK1.1质粒包含了Cas9核酸酶表达框和gRNA克隆框两个重要原件,其图谱如图1所示,大小为10 656 bp。VSP primer(CATATGCTTACCGTAACTTGAAAG)序列位于载体的U6启动子区域。下游负链oligo序列即靶位点的反向互补序列,位于载体的双Bbs I酶切位点,靶位点替换双Bbs I酶切位点。载体经酶切后,获得10 kb左右片段(图2)。

表1 sgRNA靶位点序列及靶标寡核苷酸序列Table1 sgRNAs and target oligonucleotide sequences

图1 pGK1.1质粒图谱Fig.1 The pGK1.1 plasmid map

图2 pGK1.1载体酶切后的电泳检测图Fig.2 The electrophoretic pattern of pGK1.1 vector digested by Bbs I
2.3 阳性重组子筛选与鉴定
使用上游引物VSP primer与下游负链oligo引物进行菌落PCR筛选,均能扩增出100 bp大小的片段,说明所检测单克隆全为阳性(图3)。进一步对扩增结果展开测序鉴定,并将测序结果与NCBI所提供的序列进行比对,结果显示测序正确,即载体构建成功。
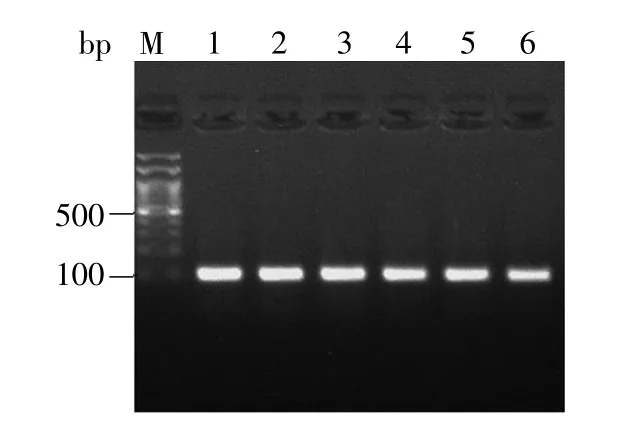
图3 菌落PCR检测电泳图Fig.3 Electrophoresis of the colony PCR products
2.4 CruiserTMEnzyme酶切筛选阳性克隆
将构建好的3个载体共同转染到SK-N-SH细胞中,制备单克隆,进行CruiserTMEnzyme酶切验证。根据扩增引物及理论敲除位点位置,扩增产物约455 bp,扩增出的目的条带大小明显变小或者可以切出条带的克隆为疑似阳性克隆。CAPNS1基因敲除SK-N-SH单克隆细胞经CruiserTMEnzyme酶切后,在400~500 bp之间出现条带,疑似为阳性克隆(图4)。

图4 CAPNS1基因的CruiserTM酶切验证Fig.4 CruiserTMEnzyme analysis of the CAPNS1 gene
2.5 单克隆测序以及TA克隆测序
将疑似阳性的SK-N-SH细胞单克隆进行单克隆测序和TA克隆测序。单克隆测序显示细胞株构建成功(图5);TA克隆测序结果比对发现两个亲本都缺失35 bp:TCAGGGACCATTTGCAGTAGTGAACTCCCAGGTGC(图6)。以上结果提示,CAPNS1基因敲除SK-N-SH细胞株构建成功。
2.6 Western-blot检测 CAPNS1、calpain1 和calpain2蛋白的表达情况
对CAPNS1基因敲除SK-N-SH稳定细胞系中的CAPNS1、calpain1和calpain2蛋白的表达情况进行Western-blot分析。结果显示,与对照组SK-N-SH细胞(CAPNS1+/+)相比,CAPNS1基因敲除SK-N-SH稳定细胞系(CAPNS1-/-)中的CAPNS1、calpain1和calpain2蛋白表达水平均明显降低(图7)。
3 讨论

图5 CAPNS1基因敲除SK-N-SH细胞系的单克隆测序Fig.5 Colony sequencing of CAPNS1 knockout SK-N-SH cells
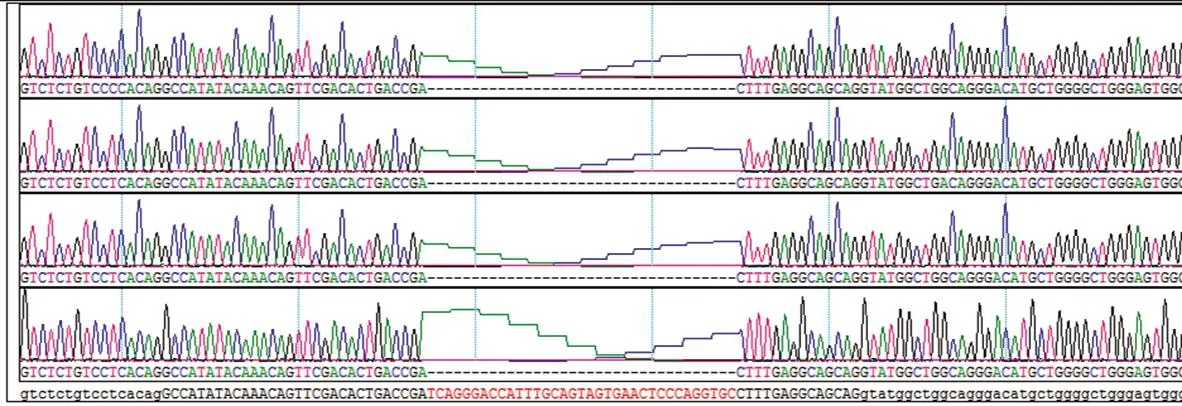
图6 CAPNS1基因敲除SK-N-SH细胞的TA克隆测序及比对Fig.6 TA cloning and alignment of CAPNS1 knockout SK-N-SH cells

图7 CAPNS1敲除后CAPNS1、calpain1和 calpain2蛋白的表达情况Fig.7 The expression of CAPNS1,calpain1 and calpain2 proteins in CAPNS1 knockout SK-N-SH cells
作为机体主要存在的细胞内蛋白质水解系统之一,calpain系统在细胞生存中具有重要作用。大量研究表明,calpain活性过渡增加或抑制与多种疾病的发生发展相关,如神经退行性疾病[12]、白内障[13]、糖尿病[14]等。CAPNS1作为 calpain1和calpain2共同拥有的小亚基结构,对calpain1和calpain2活性起着调节作用。CAPNS1由结构域Ⅴ和结构域Ⅵ两个结构域组成。结构域Ⅴ含有大量甘氨酸和疏水氨基酸,具有疏水作用;结构域Ⅵ位于C端,与大亚基结构域Ⅳ的结构相似,能与Ca2+结合,对维持calpain功能具有重要的作用。研究表明,CAPNS1基因敲除可导致calpain1及calpain2失活,影响小鼠心脏发育,从而造成其在妊娠中期死亡,提示calpain在生物的生长发育过程中起着至关重要的作用[15]。此外,有研究报道,CAPNS1对细胞自噬具有调节作用[3]。本课题组前期研究以及其他研究发现,calpain1和calpain2表达及活性的增加参与细胞自噬的调节[12],但其机制至今还不是十分清楚,需进一步的研究。
自从传统的CRISPR/Cas9系统被改造:将两个非编码RNA即crRNA和tracrRNA合成一个sgRNA,指引Cas9蛋白对靶标DNA序列进行编辑,CRISPR/Cas9系统应用越来越广泛[16]。人们利用CRISPR/Cas9基因编辑技术实现了研究不同类型基因组的目的,其不仅可作为基因编辑的工具,也可用于基因表达调控[17]。如今,CRISPR/Cas9技术已成功地被运用到人类细胞[18]、小鼠[19]、斑马鱼[20]、猪[21]等进行实验研究。但CRISPR/Cas9技术会受到PAM序列、sgRNA序列、Cas9/sgRNA的丰度等多种因素的影响,从而产生脱靶效应。对于降低CRISPR/Cas9技术的脱靶效应,我们可通过提高sgRNA序列设计的特异性、控制Cas9/sgRNA的丰度、采用双切口措施等策略实现。
本研究选择了同时包含Cas9核酸酶表达框和sgRNA克隆框的pGK1.1质粒作为载体,针对人CAPNS1基因进行敲除载体的构建。菌落PCR和测序结果显示CAPNS1基因敲除载体构建成功,进一步将其转染人成神经瘤SK-N-SH细胞,获得稳定单克隆细胞系,测序结果及Westernblot检测结果提示敲除CAPNS1基因的SK-NSH细胞系构建成功。以上实验结果为后期进一步研究calpain在自噬中的作用提供了良好的细胞模型。
猜你喜欢
环球时报(2022-09-20)2022-09-20
今日农业(2020年24期)2020-12-15
中国现代医药杂志(2020年10期)2020-12-14
中华医学图书情报杂志(2017年3期)2017-03-21
现代检验医学杂志(2016年3期)2016-11-15
小资CHIC!ELEGANCE(2015年15期)2015-09-01
医学研究杂志(2015年3期)2015-06-10
特产研究(2015年1期)2015-04-12
现代检验医学杂志(2015年4期)2015-02-06
中国药业(2014年21期)2014-05-26
