
固相萃取-气相色谱-质谱法测定3种中药材中18种多环芳烃
2022-04-07 09:07李文斌池艳艳
分析测试技术与仪器 2022年1期
李文斌,池艳艳,程 菲
(天津迪科马实验设备有限公司,天津 300499)
多环芳烃(polycyclic aromatic hydrocarbons,PAHs)是指含有两个或多个苯环以线性、角状或簇状方式排列在一起的一类化合物[1].PAHs是一种在环境中普遍存在的持久性有机污染物,由于其高脂溶性,很容易通过胃肠道被吸收,长期接触会引起免疫系统功能下降,具有强烈的致癌、致畸、致突变作用[2-3].中药在其种植生长过程中,会吸收来自空气、土壤和水中的PAHs并在体内富集积累,另外在中药的加工过程中,通过炒、烘、焙、烤等工艺也会引入PAHs从而危害人体健康[2,4].早在1979年,美国环保署(US EPA)就已经将16种PAHs列为优先控制污染物[5],韩国在2008年发布的《草药中苯并芘的规格检验方法提案》(G/TBT /N/KOR/197)中规定了生地黄和熟地黄两种草药中苯并[a]芘的最大容许量为5 μg/kg[2].欧盟2015/1933号法规规定了干草药中苯并[a]芘最大含量10.0 μg/kg,并以苯并[a]芘、苯并[a]蒽、苯并[b]荧蒽和屈4种PAHs的总量规定了干草药中PAHs最大使用限量为50.0 μg/kg[2].德国新的认证技术文件ZEK01.4-08将管控的16种PAHs提高到了18种[6].随着国际上对PAHs日益关注,为了规范中药的种植生产过程、保障人体健康、促进中药打开国际贸易市场,建立一种简便、快速测定中药材中PAHs的方法具有重要意义.
PAHs的分析方法主要包括液相色谱法(LC)[7-8]、气相色谱质谱法(GC-MS)[6,9-10]、气相色谱串联质谱法(GC-MS/MS)[11-13].早期PAHs检测常用液相串联荧光或者紫外检测器,具有较高的灵敏度,由于液相色谱主要通过保留时间定性,针对复杂的样品基质容易出现假阳性的问题.相比之下GC-MS/MS定性能力强,检出限低,适合复杂基质PAHs的分析,但同时也存在价格高昂不易推广的问题,与前两者相比GC-MS法在具备高的灵敏度和抗干扰能力的同时,价格适宜,是目前应用比较广泛的检测PAHs手段也是本文使用的检测方法.
中药材中PAHs的检测属于痕量分析,其基质复杂,含有色素、糖类化合物、萜内脂、有机酸、甾类、挥发油等多种成分.国内目前通用的食品中PAHs检测方案GB 5009.265—2016对于中药类复杂基质的检测并不完全适合.已有文献报道的针对中药PAHs前处理方法主要有固相萃取法[4,14-15]、液液萃取法[16]、凝胶渗透色谱(GPC)结合SPE法[13]、固相微萃取法[17]、浊点萃取法[18].其中固相萃取法样品用量少,净化结果明显、回收率高,集样品纯化和富集于一身、填料可选择性强,特别适合痕量成分分析,具有明显的技术优势[19],目前已广泛应用于环境、制药、食品等领域.本研究考察了鸡血藤、牡蛎、琥珀、百合、丹参、陈皮、厚朴、金银花、银杏叶、鱼腥草、平贝母、白豆蔻、枸杞等多种基质,根据前处理的难易程度及样品的洁净情况,最终选择了难、中、易三种样品,即简单基质平贝母,稍复杂基质厚朴,复杂基质丹参作为论述对象.本文采用正己烷振荡提取,通过Florisil柱和Proelut-PAH多环芳烃专用萃取柱联合使用对提取液进行净化,GC-MS检测,建立了丹参、平贝母和厚朴3种中药材中18种PAHs的分析方法.该方法操作简单、净化效率高、分析速度快,适用于类似基质中药材中PAHs的检测.
1 试验部分
1.1 仪器与试剂
GC-MS-QP2010气相色谱-质谱仪(日本岛津公司);HSC-24B氮吹仪(天津恒奥科技发展有限公司);DM-PAH多环芳烃专用色谱柱(30 m×0.25 mm×0.25 μm,迪马科技);ProElut Florisil弗罗里硅土柱(500 mg/6 mL,迪马科技);ProElut PAH多环芳烃专用萃取柱(700 mg/6 mL,迪马科技).
18种PAHs混合标准溶液,质量浓度1 000 μg/mL,溶剂为甲苯,农残级,纯度大于99.9%,迪马科技,包含萘(Naphthalene)、苊烯(Acenaphthylene)、苊(Acenaphthene)、芴(Fluorene)、菲(Phenanthrene)、蒽(Anthracene)、荧蒽(Fluoranthene)、芘(Pyrene)、屈(Chrysene)、苯并[a]蒽(Benzo [a] anthracene)、苯并[b]荧蒽(Benzo [b] fluoranthene)、苯并[k]荧蒽(Benzo [k] fluoranthene)、苯并[j]荧蒽(Benzo [j] fluoranthene)、苯并[a]芘(Benzo [a] pyrene)、苯并[e]芘(Benzo [e] pyrene)、茚并[1,2,3-c,d]芘(Indeno [1,2,3-c,d] pyrene)、二苯蒽(Dibenzo [a, h] anthracene)、苯并[g,h,i]苝(Benzo [g, h, i] perylene).正己烷、二氯甲烷、乙腈均为色谱纯(迪马科技),无水Na2SO4(天津福晨化学试剂有限公司).所使用溶剂需进行溶剂空白检测,所有玻璃器皿洗净后,用正己烷充分润洗,干燥备用.丹参、平贝母、厚朴均购自天津市某药店.
1.2 样品前处理
取中药材粉碎成粉末,过三号筛(粒径:355 μm),取约5 g,精密称定,置50 mL离心管中,加入5 g无水硫酸钠、20 mL正己烷,涡旋混合15 min,6 000 r/min离心2 min,吸取5 mL上清液待净化.取Florisil柱,依次加入6 mL二氯甲烷、5 mL正己烷活化,加入待净化液,控制流速0.5 mL/min,收集流出液1,以6 mL二氯甲烷-正己烷(体积比为1∶9)洗脱,控制流速0.5 mL/min,收集流出液2,合并流出液1、2,40 °C水浴氮吹至近干,以2 mL正己烷复溶,过PAH多环芳烃专用萃取柱(6 mL二氯甲烷、5 mL正己烷活化),3 mL正己烷淋洗,20 mL二氯甲烷洗脱,收集洗脱液在40 °C水浴氮吹至近干,加入正己烷定容到1 mL,混匀,供GC-MS分析.
1.3 标准溶液的配制
准确量取适量18种PAHs混合标准溶液,以正己烷配制成50、2.5、0.5 μg/mL的标准中间液,4 ℃冷藏保存.称取6份实际样品,经过1.2样品前处理,作为样品基质溶液,加入适量标准中间液,配制成质量浓度为5、10、20、50、100、200 μg/L的基质混合标准工作溶液.
1.4 GC-MS条件
1.4.1 色谱条件
DM-PAH色谱柱(30 m×0.25 mm×0.25 μm);载气为高纯氦气,流速2.0 mL/min;不分流进样,进样量1 μL;进样口温度:320 ℃;升温程序:初始温度65 ℃,保持0.5 min,以15 °C/min升温至220 °C,保持1 min,再以4 °C /min升温至330 °C,保持5 min.
1.4.2 质谱条件
离子源:EI源,离子源温度:230 ℃,接口温度:280 ℃,溶剂延迟时间:5 min,扫描方式:选择离子模式(SIM).18种PAHs的质谱参数如表1所列.

表1 18种PAHs质谱参数
2 结果与讨论
2.1 色谱柱的选择
根据相关文献,分析PAHs时通常使用DM-5MS柱[12]或PAH柱[4,6].本文考察DM-5MS(30 m×0.25 mm×0.35 μm)和DM-PAH(30 m×0.25 mm×0.25 μm)两种色谱柱的分离效果.结果发现,两组同分异构体苯并芘和苯并[e]芘以及苯并[k]荧蒽和苯并[j]荧蒽在DM-5MS上无法基线分离,使用DM-PAH柱经过优化升温程序,除了茚并[1,2,3-c,d]芘和二苯蒽在色谱图中存在的部分峰重叠现象,可通过它们的定性、定量离子的不同来区分,其它16种PAHs在40 min内完全分离,标准谱图如图1所示,可见各组分具有良好的分析结果.

图1 18种PAHs混合标准溶液的色谱图(SIM)
2.2 固相萃取条件的优化
2.2.1 提取溶剂的选择
样品的提取是样品前处理的重要环节,合适的提取溶剂及提取方法会直接影响后期的回收率及净化效果.多环芳烃类化合物不易溶于水,易溶于多种有机溶剂,故而不选择水作为提取溶剂.考虑到中药样品成分复杂的特点,选取强极性、弱极性的乙腈、正己烷两种溶剂对样品进行提取,并考察其提取效率.当乙腈提取样品时,PAHs类化合物回收率整体偏低,且谱图中部分化合物会有明显干扰.另外,对于颜色较深的丹参样品,乙腈提取后,提取液颜色明显较深,进一步说明使用乙腈时,杂质更多的被提取出来.采用正己烷进行提取,回收率提升比较显著,而且谱图比较干净,推断是正己烷对于非极性的PAHs溶解性更强,同时中药样品中极性干扰物质正己烷不易提取出来,基于提取更多的目标化合物且提取出更少杂质的提取原则,选用正己烷作为提取溶剂.使用不同溶剂提取的回收率结果对比如图2所示.

图2 不同提取溶剂对18种PAHs回收率影响 (n=3)
2.2.2 固相萃取柱的选择
目前,常用的样品前处理净化方法有分散固相萃取(QueChERS)、固相萃取(SPE)等.QueChERS是近年来发展起来的一种样品前处理技术,具有快速、便捷等特点.但是该法存在一定的局限性,由于方法没有淋洗过程而是通过单一的吸附杂质机理进行净化,所以对于复杂化合物的前处理还存在净化效果不理想的问题.而中药样品成分复杂,通常有糖类、氨基酸、蛋白质、油脂、蜡、酶、色素、维生素、有机酸、鞣质、无机盐、挥发油、生物碱、甙类等干扰检测成分,通过填料组合吸附杂质的净化模式(QueChERS)不易去除.SPE作为经典样品前处理技术,在净化复杂样品基质面具有独特优势.
本试验最初选用PAH多环芳烃专用萃取柱进行净化,PAH多环芳烃专用萃取柱为复合柱产品,内含弱阴离子填料及分子印迹材料,可通过离子交换作用力、氢键作用力吸附样品中有机酸、碳水化合物、色素、氨基酸等杂质,同时还具有分子印迹材料对目标化合物PAHs进行选择性吸附作用,采用正己烷上样、淋洗,二氯甲烷洗脱,对于简单基质平贝母,稍复杂基质厚朴样品可达到良好的回收率和净化效果.但是对于复杂基质丹参,苯并[b]荧蒽、苯并[k]荧蒽、苯并[j]荧蒽、苯并[a]芘、苯并[e]芘几个化合物出峰位置有明显干扰,同时基线偏高.考虑到样品基质特性,丹参中含有大量丹参酮、丹参酸等干扰物质,可与极性吸附材料表面羟基形成氢键相互作用力,故而采用Florisil柱预净化的方式,即样品提取液先进行Florisil柱通过保留干扰物模式初步净化,然后再使用PAH多环芳烃专用萃取柱通过保留目标化合物模式再净化,由图3可知,除杂效果明显,化合物可准确定量.

图3 不同萃取柱净化丹参样品谱图对比
2.2.3 流速的选择
固相萃取净化过程中,流速是影响试验结果的重要因素之一.通过试验发现,在进行Florisil柱预净化时,流速对回收率影响较大,常规流速约为1 mL/min,但回收结果偏低,多数化合物回收率低于80%,降低流速至约为0.5 mL/min时,回收率明显提升,均高于80%,不同流速结果如图4所示,最终选择0.5 mL/min为Florisil柱预净化时流速.

图4 Florisil柱流速对18种PAHs回收率影响 (n=3)
2.2.4 洗脱溶剂选择
选用丹参加标样品进行提取,提取液以Florisil柱预净化,分别选用正己烷、1%二氯甲烷正己烷、5%二氯甲烷正己烷、10%二氯甲烷正己烷、20%二氯甲烷正己烷进行洗脱,流程同1.2.结果如图5所示,当采用10%、20%二氯甲烷正己烷时,18种PAHs回收率符合要求,以洗脱结果回收率高、洗脱液极性尽可能低为原则,在达到回收率要求的同时尽可能的减少洗脱下来杂质,最终选择10%二氯甲烷作为洗脱溶剂.

图5 洗脱溶剂对18种PAHs回收率影响 (n=3)
2.3 基质效应
基质效应(matrix effect, ME)是指样品中的一种或几种非待测组分对待测物浓度或质量测定准确度的影响,包括基质抑制和基质增强两种效应.中药样品基质复杂,进行样品分析时容易受到基质干扰从而影响分析结果.本文采用中药实际样品,经过1.2净化过程前处理得到基质溶液加入定量标准品与纯溶剂中加入相同浓度标准品的方式评价基质效应(ME)的影响,计算公式如下:ME=(扣除本底后基质溶液中目标物的峰面积/溶剂中相应目标物的峰面积-1)×100%,当ME大于0时为基质增强,ME小于0时为基质抑制.ME的绝对值低于20%认为不存在明显的基质效应.试验结果如图6所示,不同样品基质效应明显不同,多数化合物高于20%,因此,本试验采用样品基质溶液配制标准曲线,以消除基质效应的影响,提高分析结果的准确性.
2.4 检出限、定量限与线性范围
选取丹参、厚朴、平贝母样品基质,采用1.3方法配制不同浓度基质标准溶液,以目标物定量离子的峰面积为纵坐标(y),对应的质量浓度为横坐标(x)绘制标准曲线.各样品中18种PAHs在5~200 μg/L的质量浓度内线性关系良好,线性相关系数均大于0.998.以信噪比为3计算,检出限(LODs)为0.6~1.2 μg/kg.以信噪比为10计算,定量限(LOQs)为2.0~4.0 μg/kg.18种PAHs在各样品中标准曲线、相关系数、检出限以及定量限如表2所列.

表2 18种PAHs标准曲线、相关系数、检出限以及定量限
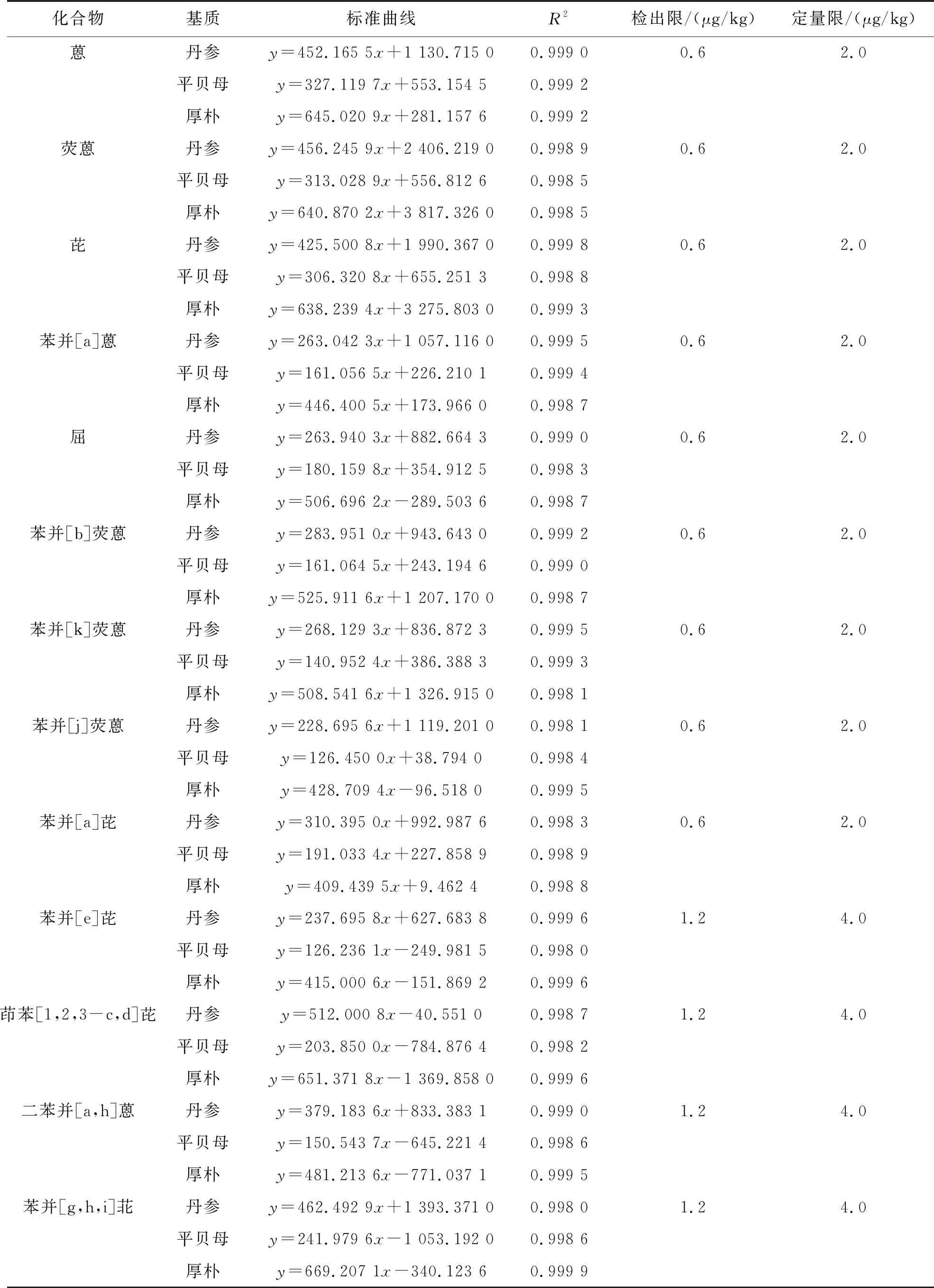
续表2
2.5 方法的回收率与精密度
取中药材粉碎成粉末,过三号筛(粒径:355 μm),取约5 g,精密称定,分别以4.0、8.0、40.0 μg/kg添加水平添加18种PAHs混合标准溶液,按照1.2方法进行处理,每个样品平行测定6次,计算加标回收率和相对标准偏差(RSD),结果如表3所列.由表3可见,丹参的回收率在81.5%~106.3%之间,相对标准偏差(RSD)为1.5%~7.8%.平贝母的加标回收率在87.1%~108.3%之间,相对标准偏差(RSD)为2.1%~8.4%.厚朴的加标回收率在82.4%~107.9%之间,相对标准偏差(RSD)为1.8%~6.9%.

表3 18种PAHs在丹参、平贝母和厚朴中加标回收率和相对标准偏差(n=6)

续表3
2.6 实际样品测定
按照上述检测方法对市售的丹参、平贝母和厚朴样品进行测定,结果如表4所列.由表4结果可见,三种药材中分别检出不同程度的PAHs,其中丹参样品中有少量芴、荧蒽和芘检出,平贝母样品中只有少量苊检出.厚朴样品中苊烯、芘、苯并[a]蒽、屈、二苯并[a, h]蒽均有检出,PAHs总检出量为293.38 μg/kg,远高于丹参和平贝母的15.24和3.55 μg/kg.

表4 丹参、平贝母和厚朴中18种PAHs残留量结果
3 结论
本研究建立了固相萃取-气相色谱质谱联用法同时测定丹参、平贝母和厚朴等3种中药材中18种PAHs的检测方法,对提取溶剂、固相萃取柱、流速、洗脱溶剂等条件进行优化.在丹参、平贝母、厚朴样品中,以不同浓度添加水平进行方法学验证,回收率及相对标准偏差符合方法学要求.18种PAHs在5~200 μg/L的质量浓度内线性良好,检出限在0.6~1.2 μg/kg之间,定量限在2.0~4.0 μg/kg之间.
该方法净化效率高,准确度、灵敏度良好,可为类似基质中药材中PAHs安全评价和质量控制提供参考.通过实际样品检测,3种药材中PAHs均有不同程度检出,应引起有关部门重视,采取相应措施,系统代码(简称“GS1统一编码”),与省财政资产云系统互联互通,使省大仪平台能获取使用省财政资产云系统的所有单位的科研仪器底数情况.截止目前,省大仪平台已集聚14 596台可共享的30万元(含)以上大型科研仪器,原值达137.95亿元.其中,国产科研仪器有3 628台,原值达34.74亿元,占比分别为24.86%、25.18%,如图2所示.
以确保中药材的安全性.重视中药材的质量管控,对于保障人体健康、促进中药现代化、走向国际贸易市场具有重要意义.
猜你喜欢
化工设计(2022年4期)2023-01-02
化学工程师(2022年3期)2022-04-19
石油炼制与化工(2022年2期)2022-02-15
绿色科技(2021年12期)2021-07-22
化工自动化及仪表(2021年3期)2021-06-04
食品工程(2020年4期)2021-01-20
酿酒科技(2019年10期)2019-11-12
电子技术与软件工程(2016年24期)2017-02-23
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
