
刺果甘草全基因组Survey及叶绿体基因组特征分析*
2022-09-29 15:19向如双段宝忠BotirKhaitovAtiatulWahab孟祥霄
世界科学技术-中医药现代化 2022年5期
向如双,孙 伟,段宝忠,王 艳,Botir Khaitov,Atia tul Wahab,孟祥霄**
(1. 大理大学药学院 大理 671000;2. 中国中医科学院中药研究所中药鉴定与安全性评估北京市重点实验室北京 100700;3. 卡拉奇大学国际化学与生命科学研究院化学研究所 卡拉奇 75270;4. 国际生物盐农业中心塔什干 100084;5. 卡拉奇大学国际化学与生命科学研究院分子医学与药物研究所 卡拉奇 75270)
甘草属GlycyrrhizaL.全球约有20 余种,我国约有8种[1],该属多数植物具有药用价值。但近年来随着生态环境的破坏及无节制的采挖,该属植物资源状况日益严峻,如何保护及高效利用甘草属药用植物资源已成为急需解决的关键问题[2]。刺果甘草G. pallidiflora主要分布于中国东北和东部地区,在阿穆尔河流域和西伯利亚也有发现,其根常用于缓解咳嗽、疼痛、溃疡和癌症等[3]。刺果甘草与2020版《中国药典》收载的甘草药材基原植物相比,其生物量较大,抗性较强,根部活性成分含量和植物生长发育具有物种的独特性[4]。
目前,有关刺果甘草的研究主要集中于化学成分[5-7]、易混伪品鉴别[8-10]、核型及染色体倍性[11-13]等方面。现代研究表明,刺果甘草化学成分有三萜皂苷类、黄酮类、香豆素类、二萜类、二苯乙烯类及多糖等成分[5]。由于形态相似,市场上刺果甘草常与乌拉尔甘草、苦参、黄芪等药材相混淆,DNA 分子标记是中药材鉴定的一种有力工具[9],当前DNA 条形码,SSR,RAPD 等技术已运用于甘草属植物的鉴定,但主要研究目标以《中国药典》中甘草药材的三个基原物种居多。有关学者开展了刺果甘草G.pallidiflora的核型研究,结果显示其体细胞染色体数目2n=16[11],与乌拉尔甘草G. uralensisFisch.、光果甘草G. glabraL.、胀果甘草G. inflataBatal.等相比,刺果甘草为最原始的一类[12]。全基因组序列的组装是研究植物代谢调控、药物活性成分积累和形成机制的重要基础[14],基因组Survey 分析可为基因组测序及组装策略提供重要参考,减少盲目性[15]。对刺果甘草叶绿体全基因组的简单重复序列、密码子偏好性、系统发育学等进行分析,有助于对刺果甘草表型产生的遗传背景及系统发育进化方面的解析。
鉴于此,本研究使用Illumina 测序技术及生物信息学方法对刺果甘草进行了基因组Survey 和叶绿体基因组特征分析,以期从基因组学层面对甘草属药用植物资源的开发、保护与利用提供理论与数据基础。
1 材料
样品的新鲜叶片采自黑龙江中医药大学(N45°43′32.71″,E126°38′55.44″),经昆明采智生物技术有限公司鉴定为刺果甘草G. pallidiflora(标本号:HBGPCGGC),新鲜叶片液氮速冻研磨后于-80℃超低温冰箱保存备用。
2 方法
2.1 基因组DNA的提取
叶片基因组DNA 使用天根植物基因组DNA 提取试剂盒(DP305)提取,DNA 浓度使用紫外分光光度计测定、完整性使用琼脂糖凝胶电泳检测。
2.2 文库构建及测序
DNA 样品检测合格后,使用超声波破碎仪(Covaris,美国)随机打断,再经末端修复、加A尾、加测序接头、纯化、PCR 扩增等步骤完成整个文库制备工作。采用Agilent 2100(安捷伦,美国)对文库的插入片段进行检测,采用Q-PCR 方法对文库的有效浓度进行准确定量,以保证文库质量[14]。文库检测合格后,通过诺禾致源科技股份有限公司高通量测序平台Illumina NovaSeq(Illumina,美国)[16]进行双末端150 bp 测序。对双端测序数据进行统计与质量评估(质量值Q20、Q30评估),采用fastp(版本:0.20.0)软件对原始数据进行过滤后得到高质量数据,原始测序数据已上传至NCBI 序列读取档案(SRA,登录号:SRR17594212)。采 用blastn(版 本:2.9.0;参 数:-evalue 1e-5 -max_target_seqs 1-bitscore 270)软件对测序reads进行NT 库比对,然后采用MEGAN(版本:6.16.4)软件进行物种分类,以判断测序数据是否存在污染,质控后数据用于基因组Survey及叶绿体全基因组分析。
2.3 19-mer分析及基因组大小估计
采用基于K-mer[17]的分析方法,对刺果甘草的基因组大小和杂合率等进行估计;采用Jellyfish(版本:2.2.10)软件,统计K-mer频数分布(K=19),获得K-mer分布图和深度估计值[15]:采用GCE[18](版本:1.0.0)软件对K-mer 频数分布数据进行分析,获得刺果甘草的基因组大小、杂合度和重复序列比例等。
2.4 叶绿体基因组的组装与注释
采用SPAdes(版本:3.14.0;参数:-k 127)软件[19]进行基因组拼接,将拼接结果与近缘参考基因组进行blastn(版本:BLAST 2.2.30+;参数:-evalue 1e-5)比对,基于比对情况并确定候选序列组装结果。采用PGA(版本:1)软件[20]对叶绿体全基因组序列进行注释并绘图,组装、注释的叶绿体基因组已上传至GenBank(登录号:MZ052084)。
2.5 叶绿体基因组重复序列和密码子偏好性分析
采用CodonW(版本:1.4.4)软件对叶绿体全基因组密码子偏好性进行分析,统计同义密码子相对使用度(Relative Synonymous Codon Usage,RSCU)的值。采用VMATCH(http://www. vmatch. de/)软 件(参 数:minimal repeat size 30 bp)分析叶绿体基因组中的散在长重复序列片段。简单重复序列(Simple Sequence Repeats, SSR)分析采用MISA(版本:1.0;参数:默认;unit size:1-8 2-4 3-4 4-3 5-3 6-3)软件分析。
2.6 叶绿体全基因组比对分析
以刺果甘草叶绿体基因组序列作为参考,采用mVISTA 对本文组装的刺果甘草G. pallidiflora(MZ052084)叶绿体基因组序列与乌拉尔甘草G.uralensis(KU862308)、光果甘草G.glabra(NC_024038)和胀果甘草G.inflata(NC_042146)的叶绿体基因组序列进行全基因组比对及差异性分析。
2.7 叶绿体基因组系统进化分析
采用MAFFT(版本:7.471)软件[21]对叶绿体基因组序列进行比对。采用RAxML[22](版本:8.2.12;核苷酸模型:GTRGAMMA;重复迭代次数:1000)软件使用最大似然法(Maximum Likelihood,ML)构建系统发育树。采用FigTree(版本:1.4.4)软件对系统发育树进行美化。
3 结果与讨论
3.1 测序数据质量评估
利用Illumina NovaSeq 平台进行测序,经过滤后,获得有效测序数据213,823,950 reads,约31.6G,原始数据及质控后数据质量Q20 值均大于90%、Q30 值均大于85%(表1),表明测序质量较好。在测序的reads中提取前50,000 条与NT 数据库进行blastn 比对,物种分类结果显示,比对上最多的是甘草属Glycyrrhiza,reads数占比对上NT库reads数的39.25%,无异常比对结果,表明基因组测序数据不存在污染。

表1 刺果甘草二代测序数据量信息统计表
3.2 19-mer分析及基因组大小估计
测序reads 的K-mer 分布结果见图1,可见分布曲线成峰情况较好,有一个主峰,K-mer深度分布主峰约为25。K-mer 分布计算结果表明,全基因组大小约为577.82 Mb,重复序列比例约53.72%,杂合度约0.31%(表2),根据估测的基因组大小,测序深度约为55X。根据基因组可以划分为微杂合基因组(0.5%≤杂合率<0.8%)、高杂合基因组(杂合率≥0.8%)以及高重复基因组(重复序列比例≥50%)[23],表明本研究中刺果甘草材料存在低杂合且重复序列较多的特点,为二倍体基因组。
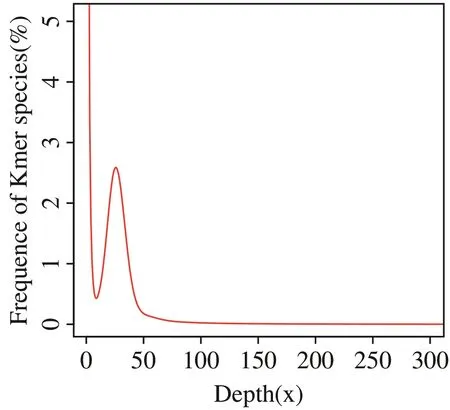
图1 K-mer=19时K-mer深度和频率分布图

表2 刺果甘草K-mer分析统计表
3.3 叶绿体基因组组装与注释
刺果甘草叶绿体基因组全长为127,267 bp,GC 含量为34.32%,其双链环状DNA 见图2,不具有典型的四分体结构,一对反向重复区(Inverted Repeat Regions,IRa 和IRb)丢失,仅含1 个大单拷贝区(Large Single Copy,LSC)和1个小单拷贝区(Small Single Copy,SSC)。经PGA 注释结果显示,其叶绿体基因组共包含110 个基因,其中76 个蛋白质编码基因,30 个tRNA 基因和4 个rRNA 基因(表3)。经与同属物种比较,结果显示刺果甘草的叶绿体基因组结构、基因功能分类与其它甘草属物种一致[24]。

图2 刺果甘草叶绿体基因组图谱

表3 刺果甘草叶绿体基因组基因列表
3.4 叶绿体基因组重复序列分析
在刺果甘草叶绿体基因组中检测到的散在长重复序列片段共有37条,包括3种类型:正向(24条)、回文(8 条)和串联(5 条),其片段的长度范围均为30-224 bp。根据刺果甘草叶绿体基因组简单重复序列(SSR)分析表明,共有243 个SSR 位点(表4),其中包括单核苷酸重复基序(141 个)、二核苷酸重复基序(86个)、三核苷酸重复基序(4 个)、四核苷酸重复基序(11个)和六核苷酸重复基序(1 个),未发现五核苷酸重复基序。在所检测的SSR 中,SSR 的类型以A/T(139 个)、AT/TA(76 个)为主,表明SSR 偏好使用A 和T 碱基。据报道SSR 在甘草属植物DNA 分子标记技术中应用较少,若将SSR 分析与多种分子技术相结合能够更好地反应甘草属植物的演化历程和进化趋势[25],为甘草属植物分子鉴定的标记开发提供基础。

表4 刺果甘草叶绿体基因组SSRs类型及数量
3.5 叶绿体基因组密码子偏好性分析
根据刺果甘草叶绿体基因组密码子RSCU 值的统计结果显示(图3),检测到共有63种密码子编码20个氨基酸,RSCU 值与编码单个氨基酸的密码子数量呈正相关,RSCU 的最小值为0.31,最大值为2.01。RSCU值最大的是UUA编码的Leu(亮氨酸)。最不常见的氨基酸是Met(蛋氨酸)和Trp(色氨酸),最常见的氨基酸是Leu(亮氨酸)、Arg(精氨酸)和Ser(丝氨酸)。在RSCU 值>1 即使用更频繁的密码子中,大多数密码子第三位均以A/U 结尾,表明刺果甘草叶绿体基因组主要偏好以第三位碱基为A 和U 为主的密码子,这与已报道的大多数药用植物的叶绿体基因密码子偏性分析的结果相似[26-27]。

图3 刺果甘草蛋白质编码基因中20个氨基酸和终止密码子的密码子含量
3.6 叶绿体基因组序列变异分析
叶绿体全基因组比对分析结果见图4,可见刺果甘草、乌拉尔甘草、光果甘草和胀果甘草叶绿体基因组序列之间相似性很高,非编码区域序列变异高于编码区域,LSC 区明显大于SSC 区。由图可见,变异较大的基因有clpP、ycf2,其他基因保守程度非常高,绝大多数基因的相似度均在90%以上。4个甘草属植物的基因间区变异大于基因区,如:psbA-trnK-UUU、trnLUAA-trnT-UGU、trnC-GCA-rpoB、 atpA-trnR-UCU、trnQ-UUG-accD、rbcL-atpB、ycf3-psaA、trnG-psbZ、atpH-atpF等,这些位点可以作为甘草属植物分子鉴定标记的候选区段。其中有4 个基因间区区域(trnLUAA-trnT-UGU、 trnC-GCA-rpoB、 atpA-trnR-UCU、trnQ-UUG-accD)和1 个蛋白编码区域(ycf2)在乌拉尔甘草、光果甘草和胀果甘草滑动窗分析中已被识别为热点区域[28]。

图4 4种甘草属植物叶绿体基因组全局比对图
3.7 叶绿体基因组系统发育分析
为确定刺果甘草的系统发育位置,使用11条叶绿体基因组序列(包括1 条豆科苜蓿属蒺藜苜蓿Medicago truncatulaGaertn.序列作为外类群,10 条豆科甘草属序列)构建ML 系统发育树,结果显示刺果甘草G.pallidiflora与其他甘草属物种聚为一支(图5),且亲缘关系与圆果甘草G.squamulosa的亲缘关系较近,研究结果与杨萍[12]的甘草核型分析结果一致。本研究的刺果甘草叶绿体基因组序列可为豆科后续开展遗传多样性研究提供重要信息。

图5 基于叶绿体全基因组序列构建ML系统进化树
4 结语
据报道,基因组Survey 分析方法已经成功应用于三岛柴胡[29]、地黄[14]、罗汉果[15]等药用植物的基因组大小测定。基于基因组Survey 分析方法来估测物种基因组大小,不仅对物种基因组测序及组装策略提供重要参考,还可以对相关蛋白组、转录组以及代谢产物研究提供理论基础[30],促进分子生物学在药用植物保护及培育优良品种方面的研究进展。叶绿体是与光合作用直接相关的细胞器,普遍存在于植物中[31],叶绿体基因组多为母系遗传、信息位点丰富、基因组成及结构相对保守[31-33],具有四分体结构,但少数植物如豆科的地车轴草Trifolium subterraneumL.、蒺藜苜蓿M.truncatula和鹰嘴豆Cicer arietinumL.等因一个反向重复区完全丢失而具有特殊的叶绿体基因组结构[31]。叶绿体基因组学研究现已广泛应用于植物物种鉴定、系统进化、遗传多样性等多个领域[34]。豆科是被子植物中三大科之一[35],目前有关豆科植物的叶绿体基因组结构研究主要集中在蝶形花亚科(Papilionoideae)[36]。
本研究利用Illumina NovaSeq 平台,完成了刺果甘草全基因组Survey 分析、叶绿体基因组组装、注释、密码子的偏好性、简单重复序列及系统发育分析等。研究结果表明刺果甘草基因组K-mer 深度分布主峰约为25,基因组大小约为577.82 Mb,基因组大小与豆科红车轴草Trifolium pratenseL.、菜豆Phaseolus vulgarisL.和绿豆Vigna radiata(L.) Wilczek 等植物较接近,符合豆科植物基因组的大小在高等植物中处于中游水平的特点[35]。K-mer 曲线服从泊松分布,杂合度约为0.31%,重复序列比例约53.72%,这与豆科沙冬青Ammopiptanthus mongolicus(Maxim.ex Kom.)Cheng f.[37]基因组特征相近,存在低杂合和重复序列较多的特点。同属的乌拉尔甘草基因组约400.95 Mb,杂合度约0.36%,重复序列比例约36.48%[38]。刺果甘草与乌拉尔甘草相比杂合度略低,更高的重复序列比例也导致了更大的基因组。刺果甘草基因组研究,有助于推动甘草属植物遗传多样性的解析,以及比较基因组和泛基因组学的研究。通过基因组、转录组、代谢组学的联合分析,可辅助解析甘草药材品质的形成机制[39]。为了更好地对刺果甘草全基因组进行序列拼接和组装,可尝试采用三代测序结合二代测序的分析策略进行基因组组装[30],本研究产生的Illumina测序数据还可用于基因组初步组装与注释等工作,解析刺果甘草的基本生物学信息,为后续开展基因克隆等分子遗传研究提供数据基础。叶绿体全基因组分析结果显示,刺果甘草叶绿体基因组具有典型的环状DNA双链结构,无反向重复(IR)区域,与已发表的豆科蝶形花亚科甘草属物种叶绿体基因组一致,都发生了IR 区域的丢失,因此豆科蝶形花亚科中的甘草属物种也可以被命名为IRLC(Inverted Repeat-Lacking Clade)分支[24,36,40];重复序列分析结果显示,刺果甘草叶绿体基因组中共检测到243个SSR,37条长重复序列,这可为甘草属植物分子标记开发、系统发育研究和相关物种鉴定提供相应的理论基础。叶绿体基因组序列变异分析结果显示,刺果甘草、乌拉尔甘草、胀果甘草和光果甘草的psbA-trnK-UUU、rbcL-atpB、ycf3-psaA、trnG-psbZ、atpH-atpF等基因间区变异较大,这些位点为甘草属植物的分子鉴定提供了新的位点资源。系统发育分析显示,刺果甘草与圆果甘草G.squamulosa的亲缘关系最近,为后续开展遗传多样性研究奠定了科学基础。
猜你喜欢
福建农业学报(2021年6期)2021-08-18
课程教育研究·学法教法研究(2019年18期)2019-10-08
少儿科技(2019年9期)2019-09-10
发明与创新·中学生(2019年6期)2019-06-26
生物学教学(2018年2期)2018-08-07
安徽农业科学(2018年1期)2018-05-14
考试周刊(2017年26期)2017-12-12
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
