
3-异硫氰酸酯氧化吲哚的合成及其在构建螺环氧化吲哚类化合物中的应用
2014-08-11 02:29赵建强袁伟成
遵义医科大学学报 2014年3期
白 玫,左 建,赵建强,袁伟成
(1. 中国科学院 成都有机化学研究所,四川 成都 610041;2. 中国科学院大学, 北京 100049;3. 遵义医学院 药学院, 贵州 遵义 563099)
螺环氧化吲哚结构单元广泛存在于天然产物中,在已发现的手性螺环氧化吲哚类化合物中,很多都具有较高的生物活性,诸如抗病毒、抗细胞毒素、抗氧化性等[1-2]。化合物Spirobrassinin和它的类似物Methoxyspirobrassinin就是一类从十字花科植物中分离出来的植物抗毒素,从结构中看,都是含有螺环氧化吲哚结构单元的生物碱,具有杀菌和抗肿瘤活性[3]。鉴于此类化合物重要的生物活性以及潜在的药用价值,因此,发展新的合成方法用于构建手性螺环氧化吲哚类化合物具有很重要的意义。
近些年来,α-异硫氰酸酯取代的酯或酰胺参与的不对称加成/环化反应已被广泛报道,该类化合物可分别经过aldol/cyclization、Mannich/cyclization和Michael/cyclization反应来构建环状的氨基硫代甲酸酯,硫脲和硫代酰胺类化合物[4-10]。结合我们课题组一直以来对氧化吲哚的研究,我们设计在氧化吲哚C3位引入强的吸电子基团-NCS,合成一类C3位异硫氰酸酯取代的氧化吲哚1。不对称催化1对亲电试剂(C=O,C=N,C=C)的加成再环化,即可构建一系列螺环氧化吲哚类化合物。
本文以商业易得的靛红为原料,经过4步,合成1-甲基-3-异硫氰酸酯氧化吲哚 1,并将其与C=O,C=N,C=C发生串联反应,构建一系列螺环氧化吲哚类化合物。
1 实验部分
1.1 仪器与试剂 BrukerAV-300型核磁共振仪;Perkin-Elmer-341自动旋光仪;Büchi B-545熔点仪;LC-20AD高效液相色谱仪;手性色谱柱(Chiralpak AD,OD,As);所用试剂均为分析纯。
1.2 化合物1的合成(见图1) ①在100 mL的烧瓶中加入6.5 g靛红A,60mL干燥的DMF,冰浴条件下,分批加入1.95 g NaH,搅拌20 min后,再加入3.1 mL碘甲烷,在50~60oC下反应3h (TLC 监测)。待反应完全后,加入60 mL水,用乙酸乙酯,合并有机相,无水Na2SO4干燥,浓缩,重结晶得6.5 g B,收率86%。②在250 mL的烧瓶中加入6.5 g B,5 g盐酸羟胺, 100 mL水,回流反应4~5 h。然后过滤产生的白色沉淀,得粗产品C(可直接用于下一步)。③将粗产品C溶解在100 mL甲醇中,加入0.6 g 10 %钯碳,然后在氢气(1 atm)氛围下,室温搅拌10 h。反应完毕后加入浓盐酸8 mL,搅拌均匀后,过滤掉钯碳,滤液浓缩得粗产品。粗产品用50 mL二氯甲烷洗涤,得7 g D,收率92%。④在250 mL的烧瓶中,加入1.7 g D,90 mL二氯
甲烷,冰浴下加入90 mL饱和碳酸氢钠水溶液,搅拌10 min后,向有机相中注入0.8 mL CSCl2,加完后迅速搅拌20 min。然后分出有机相,水相用二氯甲烷萃取,合并有机相。有机相用无水硫酸钠干燥,减压浓缩,残留混合物用柱色谱分离(正己烷/二氯甲烷 = 2:1),得1.12 g 1-甲基-3-异硫氰酸酯氧化吲哚1,收率 64%。
化合物1:1H NMR (300 MHz, CDCl3),δ(ppm): 7.44-7.42 (m, 2H), 7.18-7.13 (m, 1H), 6.88 (d,J= 7.8 Hz, 1H), 5.27 (s, 1H), 3.24 (s, 3H);13C NMR (75 MHz, CDCl3),δ(ppm): 26.7, 56.6, 108.9, 123.1, 123.5, 124.6, 130.5, 139.5, 143.4, 169.8; HRMS (ESI): Calculated for C10H8N2NaOS [M+Na]+: 227.0250, found: 227.0246。
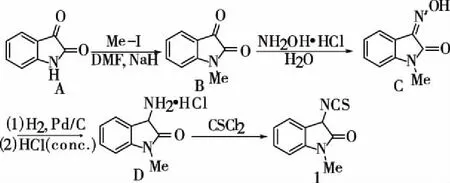
图1 合成1-甲基-3-异硫氰酸酯氧化吲哚
1.3 化合物1与C=O双键的反应 在硬质玻璃管中加入1 (0.15 mmol),Cat.A (0.1 equiv or 0.2 equiv),6 mL无水均三甲苯。混合物冷却至-40 ℃后,加入2 (2a或2b)(0.3 mmol),在-40oC反应完全。待反应完全后(TLC监测),加入1 mL浓盐酸,混合物在室温搅拌0.5 h。然后把混合物溶于乙酸乙酯,分出有机相,无水硫酸钠干燥,真空浓缩,残留物通过柱色谱纯化得化合物3/4 (见图 2)。
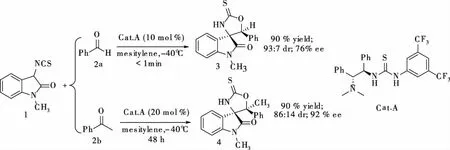
图2 化合物1与C=O双键的加成环化反应
化合物3:白色粉末,90% yield, 93:7 dr, 76% ee; [α]D20 = -3.5 (c1.00, CH2Cl2); mp 175.1~176.3oC; The ee was determined by HPLC analysis using a Chiralpak AD-H column (80/20 hexane/i-PrOH; flow rate: 1.0 mL/min; λ = 254 nm;tmajor= 6.5 min;tminor= 8.9 min);1H NMR (300 MHz, DMSO-d6),δ(ppm): 10.56 (br s, 1H), 7.81 (d,J= 7.2 Hz, 1H), 7.49-7.44 (m,1H), 7.33-7.22 (m, 4H), 7.01-6.94 (m, 3H), 6.27 (s, 1H), 2.78 (s, 3H);13C NMR (75 MHz, DMSO-d6),δ(ppm): 25.9, 70.7, 88.8, 109.2, 123.4, 123.9, 124.9, 125.4, 128.2, 129.0, 131.1, 131.8, 143.9, 171.6, 189.7; HRMS (ESI): calculated for C17H14N2NaO2S [M+Na]+:333.0668,found:333.0678。
化合物4:白色粉末, 90% yield, 86:14 dr, 92% ee; [α]D20 = +78.4 (c 0.60, CH2Cl2); mp. 210~212 ℃; The ee was determined by HPLC analysis using a Chiralpak AD-H column (80/20 hexane/i-PrOH; flow rate: 1.0 mL/min; λ = 254 nm;tmajor= 7.7 min;tminor= 12 min);1H NMR (300 MHz, DMSO-d6),δ(ppm): 10.47 (br s, 1H), 7.76 (d,J= 7.2 Hz, 1H), 7.54-7.49 (m, 1H), 7.32-7.18 (m, 4H), 7.08-7.01 (m, 1H), 6.62-6.60 (m, 2H), 2.71 (s, 3H), 1.63 (s, 3H);13C NMR (75 MHz, DMSO-d6),δ(ppm): 25.7,26.7, 72.6, 92.5, 109.4, 121.8, 122.7, 123.6, 125.6, 126.7, 128.0, 128.5, 130.4, 131.2, 138.6, 144.4,172.8, 188.8; HRMS (ESI): Calculated for C18H16N2NaO2S [M+Na]+: 347.0825, found: 347.0818。
1.4 化合物1与C=N双键的反应 在硬质玻璃管中加入1 (0.11 mmol),5 (0.11 mmol),加入3 mL二氯甲烷,然后加入NEt3(0.01 equiv), 混合物在室温搅拌12 min,反应完全后,通过柱色谱分离得化合物6 (见图3)。

图3 化合物1与C=N双键的加成环化反应
化合物6:黄色固体, mp 329.3~330.4 ℃, yield 91%, d.r. 99:1.1H NMR (300 MHz, DMSO-d6),δ(ppm): 10.37 (s, 1H), 9.30 (s, 1H), 7.32-7.30 (m, 1H), 7.24-7.22 (m, 1H), 7.15-7.04 (m, 4H), 6.94-6.88 (m, 3H), 6.82-6.79 (m, 2H), 6.58-6.56 (m, 1H), 3.68 (s, 3H), 3.01 (s, 3H);13C NMR (75 MHz, DMSO-d6),δ(ppm): 26.4, 55.2, 70.5, 78.1, 108.8, 110.2, 113.8, 121.5, 121.9, 122.6, 123.3, 125.4, 126.4, 130.0, 130.5, 130.7, 130.9, 141.8, 143.7, 158.5, 171.1, 171.6, 186.9; HRMS (ESI): Calculated for C25H20N4NaO3S [M+Na]+: 479.1148, found: 479.1149。
1.5 化合物1与不饱和C=C双键的反应 在硬质玻璃管中加入不饱和双键化合物7/8/9/10 (0.12 mmol),Cat B (0.1 equiv)或者Cat C (0.1 equiv),加入溶剂,在指定的温度下搅拌5 min后,再加入1 (0.1 mmol), 混合物继续在指定温度下搅拌(TLC监测),待反应完全后通过柱色谱分离得化合物11/12/13/14。
化合物11:白色固体; yield 99%; dr >99:1, >99% ee, [α]D20=+167.7 (c 1.01, CHCl3); m.p. 245.1~246.3 °C. The ee was determined by HPLC analysis using a Chiralpak AD-H column (70/30 hexane/i-PrOH; flow rate: 1.0 mL/min; λ = 254 nm;tmajor= 10.23 min,tminor= 7.62 min);1H NMR (300 MHz, CDCl3), δ (ppm): 3.12 (s, 3H), 4.71 (s, 1H), 6.69 (d,J= 7.5 Hz, 1H), 6.97-7.09 (m, 5H), 7.26-7.34 (m, 2H), 7.49-7.54 (m, 2H), 7.59-7.62 (m, 1H), 8.07-8.09 (m, 2H), 8.32 (d,J= 7.5 Hz, 1H), 8.55 (s, 1H);13C NMR (75 MHz, CDCl3), δ (ppm): 26.9, 59.2, 74.5, 85.6, 108.9, 123.4, 124.6, 125.1, 127.7, 128.0, 128.4, 128.6, 128.8, 129.2, 130.6, 130.8, 133.4, 143.0, 163.5, 173.0, 173.4, 198.7. HRMS (ESI) Calcd. for C26H19N3NaO3S [M+Na]+: 476.1039; found: 476.1043。
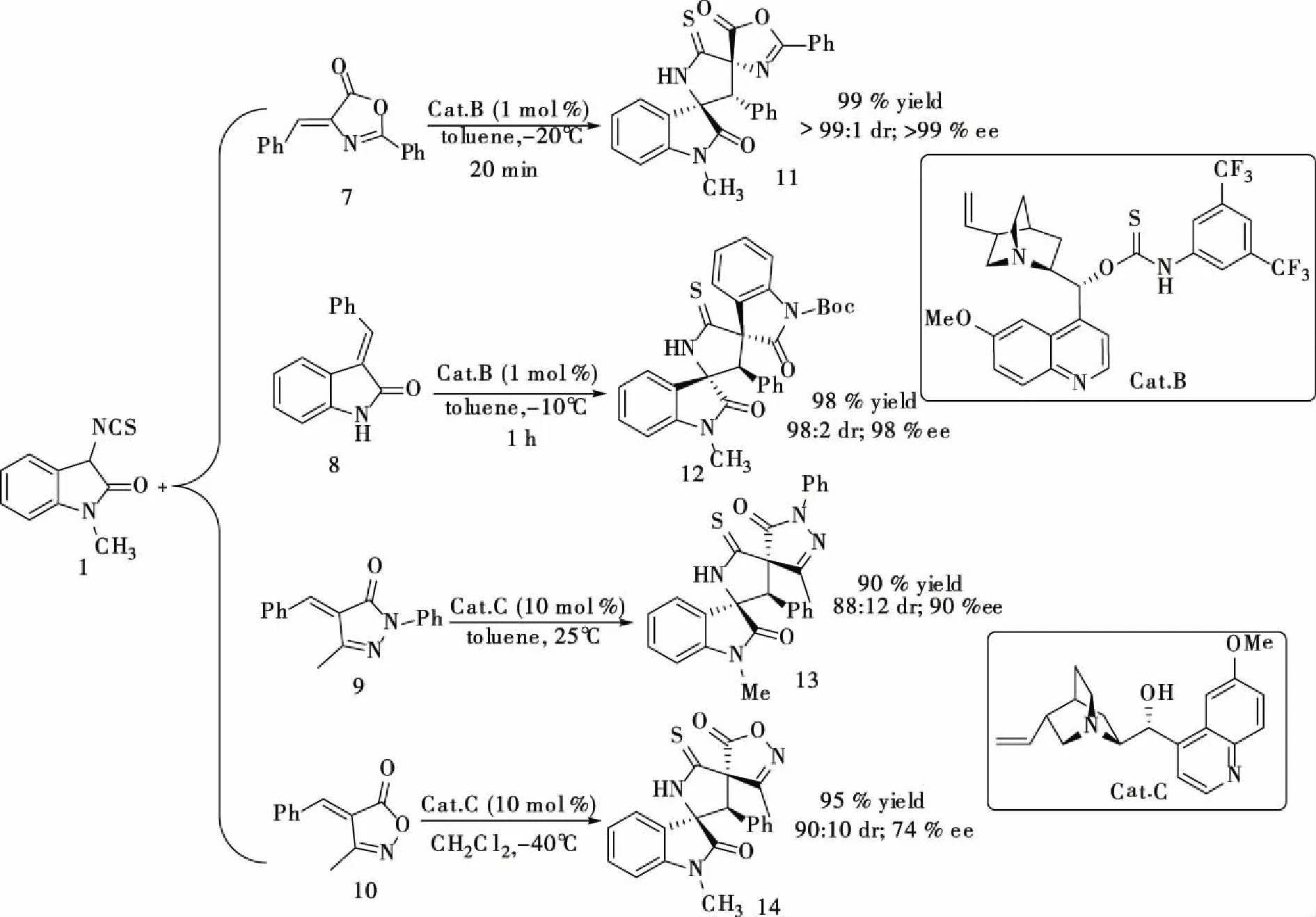
图4 化合物1与C=C双键的加成环化反应
化合物12:白色固体; yield 98%; dr 98:2, 98% ee; [α]D20=+149.6(c 0.96,CHCl3);m.p.233.3~234.1 ℃.The ee was determined by HPLC analysis using a Chiralpak OD-H column (85/15 hexane/EtOH; flow rate: 1.0 mL/min; λ = 254 nm;tmajor= 9.67 min,tminor= 18.04 min);1H NMR (300 MHz,CDCl3), δ (ppm): 1.59 (s, 9H), 3.14 (s, 3H), 5.19 (s, 1H), 6.72-6.94 (m, 6H), 7.21-7.34 (m, 4H), 7.74-7.85 (m, 3H), 8.91 (s, 1H);13C NMR (75 MHz, CDCl3), δ (ppm): 27.0, 27.9, 61.6, 72.6, 74.5, 84.5, 109.3, 115.4, 123.0, 124.2, 125.4, 125.6, 125.9, 126.7, 127.7, 127.9, 129.6, 129.8, 130.8, 131.0, 140.8, 143.5, 148.6, 173.0, 175.7, 202.9. HRMS (ESI) Calcd. for C30H27N3NaO4S [M+Na]+: 548.1614; found: 548.1638。
化合物13:白色固体, 94% yield; 88:12 dr, 90% ee; [α]D20=+17.9 (c1.07, CHCl3); mp 139.2~140.5 ℃;The ee was determined by HPLC analysis using a Chiralpak AD-H column (i-PrOH/hexane = 30/70, flow rate 1.0 mL/min,λ= 254 nm, major diastereomer:tmajor= 5.7 min,tminor= 18.4 min);1H NMR (300 MHz, CDCl3)δ(ppm): (major) 2.58 (s, 3H), 3.12 (s, 3H), 4.93 (s, 1H), 6.79 (d,J= 7.8 Hz, 1H), 6.94 (d,J= 6.9 Hz, 2H), 7.13-7.22 (m, 5H), 7.36-7.41 (m, 3H), 7.72 (d,J= 7.5 Hz, 1H), 7.88 (d,J= 7.8 Hz, 2H), 8.71 (s, 1H);13C NMR (75 MHz, CDCl3)δ(major) 16.9, 26.6, 61.3, 73.5, 77.5, 108.9, 119.2, 124.2, 124.4, 125.4, 126.3, 127.8, 128.4, 128.5, 128.8, 128.9, 129.6, 130.8, 131.3, 137.5, 143.1, 158.7, 171.3, 173.3, 197.9; HRMS (ESI): Calculated for C27H22N4NaO2S [M+Na]+: 489.1356, found: 489.1356。
化合物14:白色固体, 95% yield; 90:10 dr, 74% ee; [α]D20=-1.1 (c1.10, CHCl3); mp 110.2-111.7℃;the ee was determined by HPLC analysis using a Chiralpak AS-H column, (i-PrOH/hexane = 50/50, flow rate 1.0 mL/min,λ= 254 nm, major diastereomer:tmajor= 13.3 min,tminor= 39.2 min);1H NMR (300 MHz, CDCl3),δ(ppm): (major) 2.43 (s, 3H), 3.13 (s, 3H), 4.75 (s, 1H), 6.81 (d,J= 7.8 Hz, 1H), 6.92 (d,J= 7.2 Hz, 2H), 7.08-7.23 (m, 4H), 7.35-7.43 (m, 1H), 7.63 (d,J= 7.2 Hz, 1H), 8.56 (br s, 1H);13C NMR (75 MHz, CDCl3),δ(ppm): (major) 14.8, 26.7, 61.5, 73.5, 74.1, 109.3, 124.2, 124.4, 125.3, 127.7, 128.8, 129.0, 129.2, 129.4, 129.5, 131.7, 143.1, 164.9, 172.9, 176.5, 195.3; HRMS (ESI): Calculated for C21H17N3NaO3S [M+Na]+: 414.0883, found: 414.0883。
2 结果
以靛红为原料,经过4步反应,以总收率51%得到了1-甲基-3-异硫氰酸酯氧化吲哚 1 (见图 1)。从反应的结果可以看出,异硫氰酸酯氧化吲哚具有很高的活性,在有机小分子催化剂的作用下,可以分别与C=O (见图 2), C=N (见图 3), C=C (见图 4)双键反应,高收率,高立体选择性的得到一系列螺环氧化吲哚类化合物。例如1-甲基-3-异硫氰酸酯氧化吲哚1与吖内酯7反应,只需要1 mol %催化剂就能得到99%的产率,>99:1 dr,>99% ee的螺环氧化吲哚产物11。
3 讨论
本文以中等收率合成了1-甲基-3-异硫氰酸酯氧化吲哚,并将该底物与C=O,C=N,C=C双键反应,构建了一系列螺环氧化吲哚类化合物,产物的产率高达99%,dr>99:1,ee>99%。由于很多螺环氧化吲哚类化合物都具有较高的生物活性,我们的方法为合成螺环氧化吲哚类化合物提供了新的途径,为新药筛选及其生物活性研究提供了化合物源,为发现具有重要生物活性化合物提供了一条重要途径。
[参考文献]
[1] Lin H, Danishefsky S J.Gelsemine:a thought-provoking target for total synthesis[J].Angew Chem Int Ed,2003,42(1):36-51.
[2] Marti C, Carreira E M.Construction of spiro[pyrrolidine-3,3-oxindoles]-recent applications to the synthesis of oxindole alkaloids[J].Eur J Org Chem,2003(12):2209-2219.
[3] Suchy M, Kutschy P, Monde K,et al.Synthesis,absolute configuration, and enantiomeric enrichment of a cruciferous oxindole phytoalexin, (S)-(-)-spirobrassinin, and its oxazoline analog[J]. J Org Chem,2001,66(11): 3940-3947.
[4] Willis M C,Cutting G A,Piccio V J,et al.The direct catalytic enantioselective synthesis of protected aryl β-hydroxy-α- amino Acids[J]. Angew Chem Int Ed,2005,44(10):1543-1545.
[5] Jiang X, Cao Y, Wang Y,et al.A unique approach to the concise synthesis of Highly optically active spirooxazolines and the discovery of a more potent oxindole-type phytoalexin analogue[J]. J Am Chem Soc,2010,132(43):15328-15333.
[6] Cutting G A, Stainforth N E, John M P,et al.Direct catalytic enantioselective mannich reactions:synthesis of protectedanti-α,β-Diamino Acids[J]. J Am Chem Soc,2007,129(35):10632-10633.
[7] Cao Y, Jiang X, Liu L,et al.Enantioselective michael/cyclization reaction sequence:scaffold-Inspired synthesis of spirooxindoles with multiple stereocenters[J]. Angew Chem Int Ed,2011,50(39):9124-9127.
[8] Tan B, Zeng X, Leong W W Y,et al.Core structure-based design of organocatalytic [3+2]-cycloaddition reactions: Highly efficient and stereo-controlled syntheses of 3,3'-pyrrolidonyl spirooxindoles[J]. Chem Eur J,2012,18(1):63-67.
[9] Wu H, Zhang L L, Tian Z Q,et al.Highly efficient enantioselective construction of bispirooxindoles containing three stereocenters through an organocatalytic cascade Michael-cyclization reaction[J].Chem Eur J,2013,9(34):1747-1753.
[10] Tan F, Lu L Q, Yang Q Q, et al.Enantioselective cascade michael addition/cyclization reactions of 3-Nitro-2H-chromenes with 3-Isothiocyanato oxindoles:efficient synthesis of functionalized polycyclic spirooxindoles[J]. Chem Eur J,2014, 2(11):3415-3420.
猜你喜欢
分子催化(2022年1期)2022-11-02
中草药(2022年9期)2022-05-06
油气·石油与天然气科学(2021年12期)2021-12-11
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
昆明医科大学学报(2020年12期)2021-01-26
合成树脂及塑料(2020年4期)2020-09-20
人物画报(2020年29期)2020-03-14
人物画报(2020年36期)2020-03-13
天然产物研究与开发(2019年1期)2019-03-01
