
[Ru(tpy)(pynp)OH2]2+的电子结构及相关性质的研究
2016-02-10 01:13王菊平张恩华
广州大学学报(自然科学版) 2016年6期
王菊平, 张恩华, 刘 玲
(广东药科大学 药科学院, 广东 广州 510006)
[Ru(tpy)(pynp)OH2]2+的电子结构及相关性质的研究
王菊平, 张恩华, 刘 玲
(广东药科大学 药科学院, 广东 广州 510006)
文章利用密度泛函理论(DFT),在B3LYP/Lanl2dZ//6-31G(d) 的水平上,对 [Ru(tpy)(pynp)OH2]2+的异构体1a(反式)和1a’(顺式)的电子结构及相关性质进行了理论研究.计算结果表明,Ru-O的键长明显比Ru-N键的要长,1a可通过水离去方式来异构化1a’;在1a’中,pynp上的N与H2O分子的H形成了氢键,此氢键影响1a’反应活性; 对于HOMO能级,1a<1a’,相反,对于LUMO能级,1a>1a’;对于1a和1a’,其吸收光谱的电子基谱带和次基谱带都可以归属为单重的金属至配体的电荷转移 (MLCT),这些计算结果可为实验合成高效DNA键合分子及高效的水氧化催化体系的设计提供理论参考.
钌多吡啶; 电子结构; 密度泛函理论
钌多吡啶类配合物具有独特的与DNA结合能力、抗肿瘤活性、激发态活性以及丰富的光物理、光化学、水氧化催化、质子耦合电子转移等性能.因此,钌多吡啶类配合物在生命科学、光学控制、环境分析、催化化学及太阳能利用等领域具有重要的应用和广阔的发展前景[1-4]. 尽管钌过渡金属多吡啶类配合物已得到广泛而深入的研究,并取得许多非常有意义的成果,但这些研究主要是停留在实验的研究层面.更重要的是,钌过渡金属多吡啶类配合物与DNA的键合强度及其在水氧化催化中的催化功能,仍存在着不少的争议和许多重要的、急需解决的问题[5-7],如钌过渡金属多吡啶类配合物与DNA的光裂解的关系问题.又如,迄今为止,水氧化催化反应一直是制约光电池发展运用的最主要因素,要解决这一难题的关键就是开发高效水氧化催化剂,并将其运用于可见光驱动水氧化体系中.钌多吡啶配合物是水氧化催化反应的重要催化剂[8-9].要真正弄清楚钌多吡啶类配合物与DNA的作用位点及解决这些存在的争议,用量子化学的方法深入到电子结构层次进行理论研究是十分必要的.本文运用DFT(Density functional theory)法,对Ru[(tpy) (pynp)OH2]2+中的2种异构体进行量子化学计算,比较了它们的几何结构、电子结构及相关性质(如光谱、轨道成分、电荷布居、配合物的稳定性等)的异同,为该类配合物的合成、功能分子设计、光催化作用机理及其与DNA的作用提供理论依据.
1 计算模型与方法
[Ru(tpy)(pynp)OH2]2+是由1个二齿配体pynp、1个三齿配体tpy以及1个H2O与Ru2+中心配位形成的类似八面体的配合物.[Ru(tpy)(pynp)OH2]2+存在对映异构体结构:反式异构体1a,顺式异构体为1a’.其结构见图1.在光照条件下,1a能够发生异构化而转换成1a’, 但在加热条件下不能转换.顺式异构体1a’的配体pynp上未配位的N原子能与配位的H2O在空间上距离较近,能形成氢键.笔者推测,此氢键的存在,将会导致1a 和1a’ 的电子结构及相关性质有较大的区别.本文使用Gaussian 09程序包[10],利用DFT(B3LYP)法,对于[Ru(tpy)(pynp)OH2]2+中的2种异构体进行了全几何优化计算.杂化的B3LYP方法已证实对该类钌配合物的计算是合适的[11-14],因此,本论文结果是可靠的,其相关性质的预测是合理的.在计算中,对于Ru2+中心,使用了LANL2DZ基组;对于C、H、N、O原子,笔者使用了6-31G(d)基组.全部计算在超级计算机上完成.为了方便讨论,笔者对一些重要原子进行了编号,见图1.
2 结果与讨论
2.1 1a和1a’的配位键长的比较
计算得到的1a和1a’的主要配位键长列在表1中.
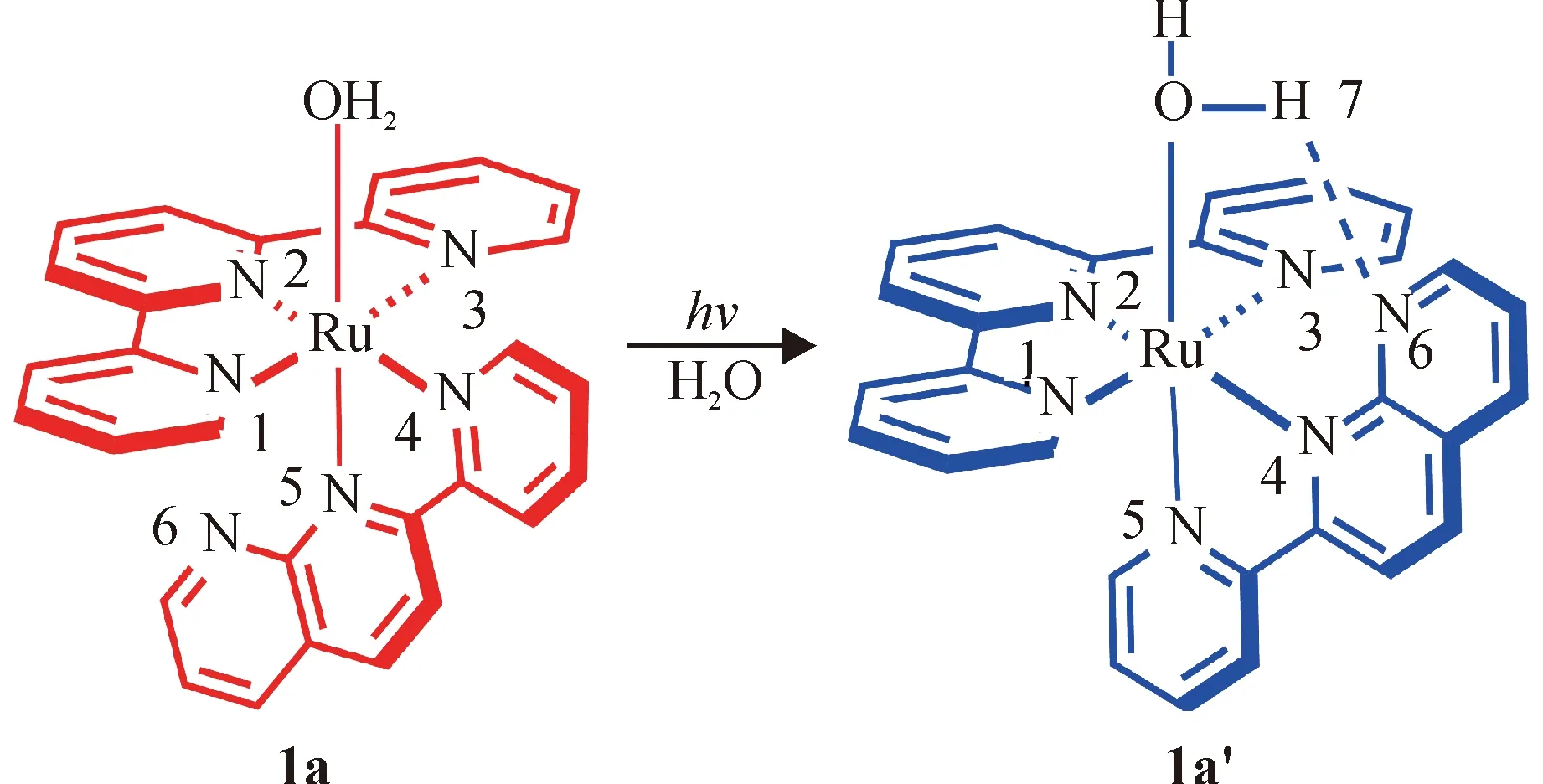
图1 1a和1a’的计算模型和原子编号
Fig.1 Calculation models and atom labels of complexes 1a and 1a’
表1 配合物1a和1a’的主要配位键长
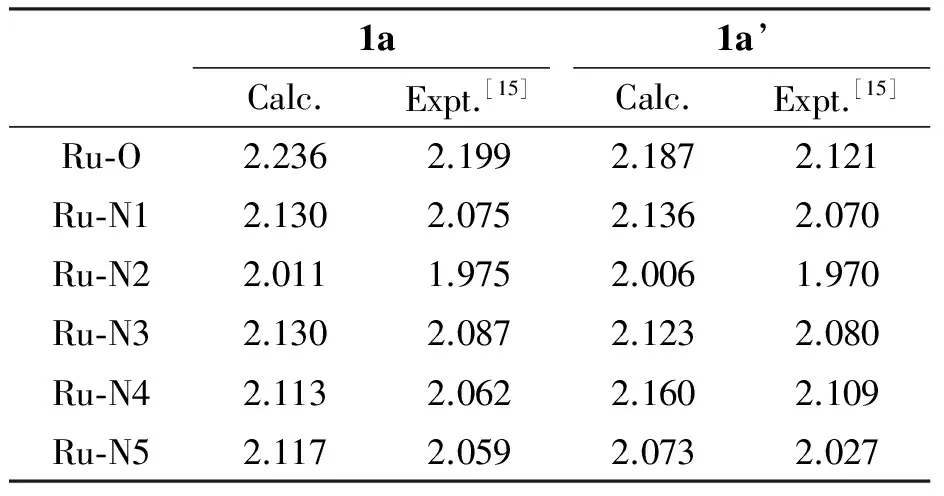
Table 1 Main coordination bond lengths of complexes 1a and 1a’ /Å
配位键长的大小反映了配位键的相对强弱,配位键越短,配位越稳定.由表1可见,计算结果与实验结果偏差很小,在2%以内. 这也反映了本文采用的计算方法和基组对所研究的体系是合适的.所有计算的配位键长比实验键长[11]仅略微偏长,而且变化趋势完全一致.以Ru-N1键为例,对于1a配合物,其计算值和实验值分别为2.130 Å和2.075 Å;而对于1a’,其计算值分别为2.187 Å和2.121 Å.计算值略大于实验值.而且对Ru-N1计算值来说,1a>1a’ ;对Ru-N1实验值,也有1a>1a’,即计算值的变化趋势与实验值的变化趋势完全一致.1a和1a’配合物配位键长主要呈现以下规律.
(1)Ru-O配位键的键长明显比所有Ru-N配位键要长,如对于配合物1a,Ru-O的计算键长为2.236 Å,比Ru-N1(2.130 Å)、 Ru-N2(2.011 Å)、Ru-N3(2.130 Å)、Ru-N4(2.113 Å)、Ru-N5(2.117 Å)都要长.这主要是由于O与N属于同一周期,相对于N原子来说,O原子对其外层电子的吸引力大,导致O原子上孤对电子与Ru2+的配位能力比N原子要弱,进而导致其与Ru2+中心形成的Ru-O配位键较弱,键长比Ru-N键长.
(2)计算的1a的Ru-O键长较1a’长0.048 1 Å (0.048 1 Å = 2.236 Å-2.187 Å),故1a的配位H2O分子更易于离去,发生异构化,转换成1a’.这也是在光照条件下,1a可以转化为1a’的原因之一.
(3)对于1a和1a’, Ru-N2 的键长都是Ru-N键中最短的.这主要是与之相邻的2个吡啶环的影响,使得N2原子的配位能力大大强于其他N配位原子.
(4)对于1a,pynp配体的Ru-N4与Ru-N5的键长几乎是相等的; 而对于1a’, Ru-N4的键长明显比Ru-N5长.这是由于在1a’中,pynp上的N与H2O分子的H形成了氢键(O-N6…H7),而且,从其结构图明显可见,pynp在配合物1a’中的空间位阻大于其在1a中的,这两方面共同导致了Ru-N4的键长拉长.值得注意的是,1a’的N6…H7的距离为1.690 Å,这意味着N6与H7之间确实存在强的氢键,如此强的氢键对1a’的电子结构和反应活性将有明显的影响.
2.2 1a和1a’的前线分子轨道能量、成分及相应性质的比较
占据的前沿分子轨道能量与光电子能谱相联系,而前沿分子轨道的能量差与可见紫外光谱相联系.尤其是HOMO、LUMO轨道能量及成分,它们在该类配合物与DNA结合及水催化氧化时起着关键性的作用.计算得到的一些前沿分子轨道能量、相应的能量差及轨道对称性列于表2 中.为了简明比较,做出与光谱关系最密切的HOMO、HOMO-1、HOMO-2、LUMO、LUMO-1和LUMO-2的能级图,而且为了进一步探讨前线分子轨道组成对1a和1a’反应性质的影响,基于计算结果,用Gaussview 5.0软件做出其轨道立体图,见图2.
表2 配合物1a和1a’的重要前沿分子轨道能量及HOMO-LUMO能隙差
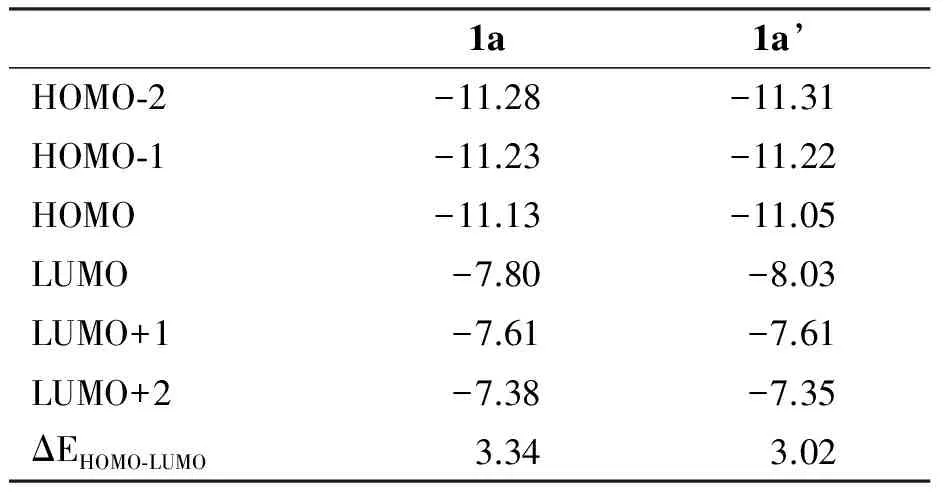
Table 2 Energies of important frontier molecular orbitals and energy gap of HOMO-LUMO /eV
表2和图2可见, 1a与1a’ 的前线轨道主要呈现出如下特点:
(1) 从能量上分析, 1a(HOMO)<1a’(HOMO);相反,1a(LUMO)<1a’(LUMO).根据前沿分子轨道理论,
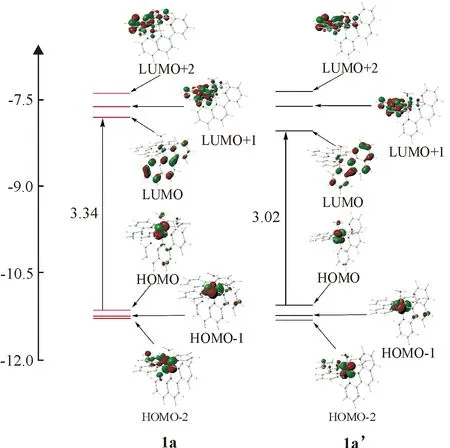
图2 配合物1a和1a’的前线轨道能量及跃迁示意图
Fig.2 Schematic representation of frontier molecular orbital energies and transitions
当分子之间通过轨道相互作用反应时,如果一个分子的HOMO轨道能量较高,而另一分子的LUMO轨道较低,则对反应进行有利,因为这样便于电子从一个分子的HOMO转移到另一个分子的LUMO,从而减少反应系统的总能量.因此,当它们与DNA相互作用时,由于DNA碱基对的前线轨道能量远高于钌配合物的前线轨道能量,甚至其HOMO能量比钌配合物的LUMO能量还要高,由此可推测,在DNA介导的电子转移过程中,1a’相对于1a来说,可作为一种更好的电子授体(因其HOMO轨道能量较高);同时,当它们以插入模式与DNA结合时(DNA的碱基对与配体通过Л电子堆积相互作用),比较容易接受从DNA的碱基对转移来的电子(因其空轨道能量较低).另外,从水氧化催化方面来说,1a’更容易失去电子,同时由于1a’的LUMO的能量低,也易于得到电子.即相对于配合物1a,1a’有更小的氧化还原电势.
(2)对于1a和1a’,HOMO、HOMO-1、HOMO-2的电子云都主要位于金属中心Ru2+的d轨道上,具有d轨道特征,因此1a和1a’参与反应时,主要是Ru2+失去电子.需要注意的是,两异构体的HOMO轨道成分含有少量O原子轨道,而H7直接与O原子相连,因此,在水氧化催化过程中,1a’配体H2O分子与N6之间的氢键将影响其催化活性;而它们的空轨道LUMO、LUMO-1和LUMO-2的电子云主要聚集在pynp或tpy配体上,具有p轨道特征.根据这些占据轨道和空轨道的电子云分布特点,可以合理预测,对于1a和1a’,它们吸收光谱的电子基谱带和次基谱带都将有明显的金属至配体的电荷转移(MLCT)特征.这种特征的光谱在钌配合物光裂解DNA时起着非常重要的作用: 1a和1a’的配体分别与DNA中的碱基相结合.在光照条件下,钌配合物基态上的1个电子,从金属中心转移到激发态的配体上(MLCT),导致金属中心Ru的化合价从+II价升高到+III价.而激发态上的电子与溶液中的氧分子(O2)则发生交换转移, 生成超氧阴离子自由基(O2·-)、单线态氧(1O2)或羟基自由基(OH·)活性氧化物种, 从而可导致DNA双链的氧化切割(即DNA光裂解).
另外,计算结果表明,1a和1a’的E(SCF)能量分别为-1 577.508 2 a.u.和 -1 577.527 0 a.u,也就是说,1a’的能量比1a的能量高12 kcal/mol, 根据能量越低越稳定原理,可以推测异构体1a’较1a更为稳定,这主要是由于1a’存在分子内氢键,降低了配合物的能量.这也与相关实验结果一致:在光照条件下,1a可以转化为1a’, 但 1a’不能转化为1a.
2.3 1a和1a’原子的净电荷布居比较
通过自然轨道分布分析(NPA)的计算,可得到1a和1a’的原子净电荷布居.在表3中,列出了与配合物反应活性紧密相关的原子的净电荷数值.

表3 1a和1a’主要原子的静电荷布居
由表3可知,1a和1a’钌上的正电荷分别为0.300 5和0.294 0,即1a的钌带有更多的正电荷,表明1a的氧化性较高,易发生质子耦合电子转移(PCET).而1a’中O原子上聚集的负电荷较1a多,主要是由于1a’中的H2O配体上的H7与N6形成了氢键(图1),使得氢原子上的电子云偏向氧原子,氧原子上的负电荷增加.对于1a和1a’, N2原子上聚集的负电荷是最少的,说明N2原子提供了较多的电子给中心离子钌,它们之间的配位更牢靠,这与Ru-N2的键长是所有N配位键中最短的是一致的.同时,由于N2原子提供更多的负电荷与中心离子配位,从而引起其对位的N4原子与钌中心的配位减弱,计算结果表明,1a中N4的净电荷为-0.398 8,在1a’中N4的净电荷为-0.411 3. 1a异构化为1a’时,1a 中pynp配体的角度会有略微的改变,且1a中的Ru-N4配位键相对较弱,将有利于1a向1a’转化.
3 结 论
通过对 [Ru(tpy)(pynp)OH2]2+的异构体1a(反式)和 1a’(顺式)的电子结构及相关性质理论计算,可得出如下主要结论:①Ru-O的键长明显比Ru-N键的要长,导致1a可通过先失水的配位,再在反向位置配上水的方式来转化为 1a’;②在1a’中,pynp上的N与配位H2O分子上的H形成了氢键,影响1a’反应活性和稳定性,1a’比1a更为稳定;③对于HOMO能级,1a<1a’,相反,对于LUMO能级,1a>1a’, 因此,1a’能更好的与DNA结合;④对于1a和1a’,占据的电子云都主要位于金属中心Ru2+d轨道上,具有d轨道特征,而它们的空轨道主要位于pynp或tpy配体上,具有p轨道特征,它们吸收光谱的电子基谱带和次基谱带都将有明显的金属至配体的电荷转移(MLCT)特征.以上计算结果合理的解释了1a和1a’结构和性质的差异,并对其与DNA键合强度、紫外可见光谱的性质及水氧化催化功能提供了合理的预测,这些有价值的信息可为实验上设计与合成高效DNA键合分子和高效的水氧化催化剂提供理论参考.
【责任编辑: 陈 钢】
Theoretical study on the electronic structures and related properties of [Ru(tpy)(pynp)OH2]2+
WANG Ju-ping, ZHANG En-hua, LIU Lin
(School of Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, China)
A detailed theoretical study on the electronic structures and related properties of [Ru(tpy)(pynp)OH2]2+isomers 1a (Trans) and 1a’ (cis) are carried out by using the DFT method at the B3LYP/LanL2DZ//6-31G(d) level of theory. Our calculations show that, for both complexes 1a and 1a’, the bond length of Ru-O is obviously longer than that of Ru-N, indicating that 1a can be converted into 1a’ via H2O dissociation manner; a hydrogen-bond is formed between N atom of pynp and H atom of H2O, which would greatly influences the reactivity of 1a’; the HOMO energy of 1a is lower than that of 1a’ but there is an inverse trend for the LUMO energy; for both 1a and 1a’,their electronic ground bands and the next ground bands in absorption spectra are all assigned to singlet metal-to-ligand charge-transfer(MLCT). These computational results provide some valuable information for experimental synthesis and design of high effective DNA binding agents and water-oxidation catalyst.
Ru polypyridyl complexes; electronic structures; DFT
2016-06-20;
2016-09-08
广东省医学科学技术研究基金资助项目(A2015556)
王菊平(1975-),女,讲师,博士. E-mail:jupingwang@gdpu.edu.cn
1671- 4229(2016)06-0046-05
TQ 426
猜你喜欢
高中数理化(2022年14期)2022-08-15
波谱学杂志(2021年3期)2021-09-07
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
青岛大学学报(工程技术版)(2019年2期)2019-09-10
原子与分子物理学报(2019年5期)2019-04-28
当代陕西(2019年6期)2019-04-17
中学化学(2015年12期)2016-01-19
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
外语学刊(2014年3期)2014-12-03
