
HPLC-ELSD法测定氨糖美辛肠溶胶囊中盐酸氨基葡萄糖及相关物质
2016-07-24 17:32王太亮王子秦
中国生化药物杂志 2016年6期
王太亮,王子秦
(1.河北省唐山市食品药品检验中心,河北 唐山 063000;2.上海师范大学,上海 200235)
HPLC-ELSD法测定氨糖美辛肠溶胶囊中盐酸氨基葡萄糖及相关物质
王太亮1Δ,王子秦2
(1.河北省唐山市食品药品检验中心,河北 唐山 063000;2.上海师范大学,上海 200235)
目的 建立高效液相色谱法-蒸发光散射检测法(high performance liquid chromatography-evaporative light scattering detector,HPLC-ELSD)法测定氨糖美辛肠溶胶囊中盐酸氨基葡萄糖的含量及其有关物质。方法 采用Inertsil C8色谱柱(250 mm×4.6 mm,5 μm),以乙腈-水(70:30)为流动相,流速为1.0 mL/min,柱温为25 ℃。结果 在选定的色谱条件下, 盐酸氨基葡萄糖进样量在25~312.5 μg/mL范围内与峰面积线性关系良好r=0.9991(n=6)。加样回收率为98.0%,RSD为1.3%(n=5);盐酸氨基葡萄糖对酸稳定,碱、热、光不稳定。结论 方法准确、快捷、重复性好,可用于含量的测定及其有关物质的检查。
盐酸氨基葡萄糖;HPLC-ELSD;含量测定;有关物质
盐酸氨基葡萄糖(分子式C6H13NO5·HCl)是甲壳素降解产物[1]。临床上,盐酸氨基葡萄糖具有多种生物活性,是预防和治疗骨关节炎疾病的特异性生化药物。
目前常用的检测方法有:紫外-可见分光光度法[2-3]、高效液相色谱-紫外检测法[4-5]、柱前衍生化-高效液相色谱法[6]和高效液相色谱-蒸发光散射检测法[7-9]。国家食品药品监督管理局标准YBH07802008中盐酸氨基葡萄糖的含量采用Elson-Morgan比色法测定,样品处理需衍生化反应,操作繁琐,且样品溶液不稳定,溶液颜色随时间变化而变化,定量重复性较差。高效液相色谱-紫外检测法所用测定波长为195 nm,恰处于乙腈的末端吸收,影响测定结果。柱前衍生化-高效液相色谱法操作复杂,检测周期长,衍生化产物不稳定,衍生化试剂产生的信噪比较大。
采用HPLC-ELSD法测定,专属性较强,出峰快,缩短分析时间,降低成本,灵敏度高、准确。
1 材料与方法
1.1 材料
1.1.1 药品与试剂:氨糖美辛肠溶胶囊(葵花药业集团衡水得菲尔有限公司,批号:20150805、20150806、20150807、201508011、201508012、20150801、20150912及20151103),盐酸氨基葡萄糖对照品(中国食品药品检定研究院, 批号: 140649-201505,供含量测定,105 ℃干燥至恒重);乙腈为色谱纯(美国)。
1.1.2 仪器:Waters e2695高效液相色谱仪(美国),在线脱气、自动进样、奥泰蒸发光散射检测器(美国)、Empower工作站,KQ-250E超声波清洗仪(江苏昆山),Sartorius BP211电子天平(德国)。
1.2 方法
1.2.1 色谱条件:色谱柱:规格型号:Inertsil C8柱(250 mm×4.6 mm , 5 μm) ;流动相:乙腈:水(70:30);流速:1.0 mL/min;柱温: 25 ℃;漂移管温度90 ℃,雾化器空气流速3.0 L/min。
1.2.2 溶液的制备
① 对照品溶液制备:精密称取盐酸氨基葡萄糖对照品12.50 mg,置100 mL容量瓶中,加水溶解并稀释至刻度,摇匀,即得。浓度为0.125 mg/mL。
② 样品溶液制备:取本品20粒,内容物精密称定,计算平均装量。混匀研细,取细粉适量(约相当于盐酸氨基葡萄糖12.5 mg),精密称定,置100 mL容量瓶中,加水65 mL超声处理(功率250 W,频率40 kHz)10 min,加水稀释至刻度,摇匀,作为供试品溶液。
③ 阴性样品溶液的制备:处方中不含盐酸氨基葡萄糖,其余同样品,溶液的制备按“②”项下样品溶液制备。
1.2.3 方法学验证
① 专属性:在上述色谱条件下,将阴性对照溶液、盐酸氨基葡萄糖对照品溶液、供试品溶液分别经0.45 μm的微孔滤膜过滤后,取10 μL进样观察色谱图。
② 线性:精密量取“1.2.2①”项下盐酸氨基葡萄糖对照品溶液,分别进样2、5、10、15、20、25 μL。峰面积(y)的对数为纵坐标,盐酸氨基葡萄糖质量浓度(x, μg/mL)的对数为横坐标作线性回归。
③ 检测限: 取“1.2.2①”项下对照品溶液,加水逐步稀释至所得到色谱图中盐酸氨基葡萄糖峰高为噪音的3倍,为最低检测限。
④ 精密度:精密吸取“1.2.2①”项下盐酸氨基葡萄糖对照品溶液10 μL进样 ,连续重复6次,测得峰面积。取同批样品6份,照“1.2.2②”项方法制备供试品溶液,按“1.2.1”项下色谱条件进样分析,分别进样10 μL,计算含量。
⑤ 加样回收:称取阴性样品细粉适量,共9份,分置25 mL量瓶中,分为3组,每组分别精密加入“1.2.2①”项下盐酸氨基葡萄糖对照品溶液12、15、18 mL,超声处理(功率250 W,频率40 kHz)10 min,加水稀释至刻度,摇匀,经0.45 μm的微孔滤膜过滤后取20 μL注入液相色谱仪测定,计算回收率。
⑥ 稳定性试验:在室温下取样品溶液于 0, 3, 6, 9, 12, 18 h分别进样分析。
⑦ 样品含量测定:取5批样品按“1.2.2②”项下制备并测定。
1.3 相关物质[10-13]
1.3.1 破坏性实验
① 酸破坏:取“1.2.2②”项下细粉适量(约相当于盐酸氨基葡萄糖12.5 mg),精密称定,置100 mL容量瓶中,加水65 mL溶解,加2 mol/L盐酸溶液1 mL,超声处理(功率250 W,频率40 kHz)10 min,用2 mol/L氢氧化钠溶液调pH值至中性,加水稀释至刻度,摇匀,滤过,即得。
② 碱破坏:取“1.2.2②”项下细粉适量(约相当于盐酸氨基葡萄糖12.5 mg),精密称定,置100 mL容量瓶中,加水65 mL溶解,加2 mol/L氢氧化钠溶液1 mL,超声处理(功率250 W,频率40 kHz)10 min,用2 mol/L盐酸溶液调pH值至中性,加水稀释至刻度,摇匀,滤过,即得。
③ 加热破坏:取“1.2.2②”项下细粉适量(约相当于盐酸氨基葡萄糖12.5 mg),精密称定,置100 mL容量瓶中,加水65 mL溶解,水浴加热15 min,放冷至室温,加水稀释至刻度,摇匀,滤过,即得。
④ 光破坏:取“1.2.2②”项下细粉适量(约相当于盐酸氨基葡萄糖12.5 mg),精密称定,置100 mL容量瓶中,加水65 mL溶解,超声处理(功率250 W,频率40 kHz)10 min,用强光(4500 LX)照射12 h以上,摇匀,滤过,即得。
1.3.2 自身对照:取“1.2.2②”项下细粉适量(约相当于盐酸氨基葡萄糖12.5 mg),精密称定,置100 mL容量瓶中,加水65 mL溶解,超声处理(功率250 W,频率40 kHz)10 min,加水稀释至刻度,摇匀,滤过,作为供试品溶液;精密量取1 mL,置100 mL量瓶中,加水稀释至刻度,作为对照溶液。
按“1.2.1”项下色谱条件,精密量取上述溶液(经0.45 μm的微孔滤膜过)各10 μL,分别注入液相色谱仪。
另取样品3批,批号:20150801、20150912、20151103,分别检测相关物质。
2 结果
2.1 方法学验证
2.1.1 专属性:阴性图谱中未出现色谱峰,样品图谱中盐酸氨基葡萄糖色谱峰保留时间与对照品色谱峰保留时间一致,并且与相邻色谱峰分离度符合要求。说明处方中其他成分对盐酸氨基葡萄糖检测无干扰。见图1。
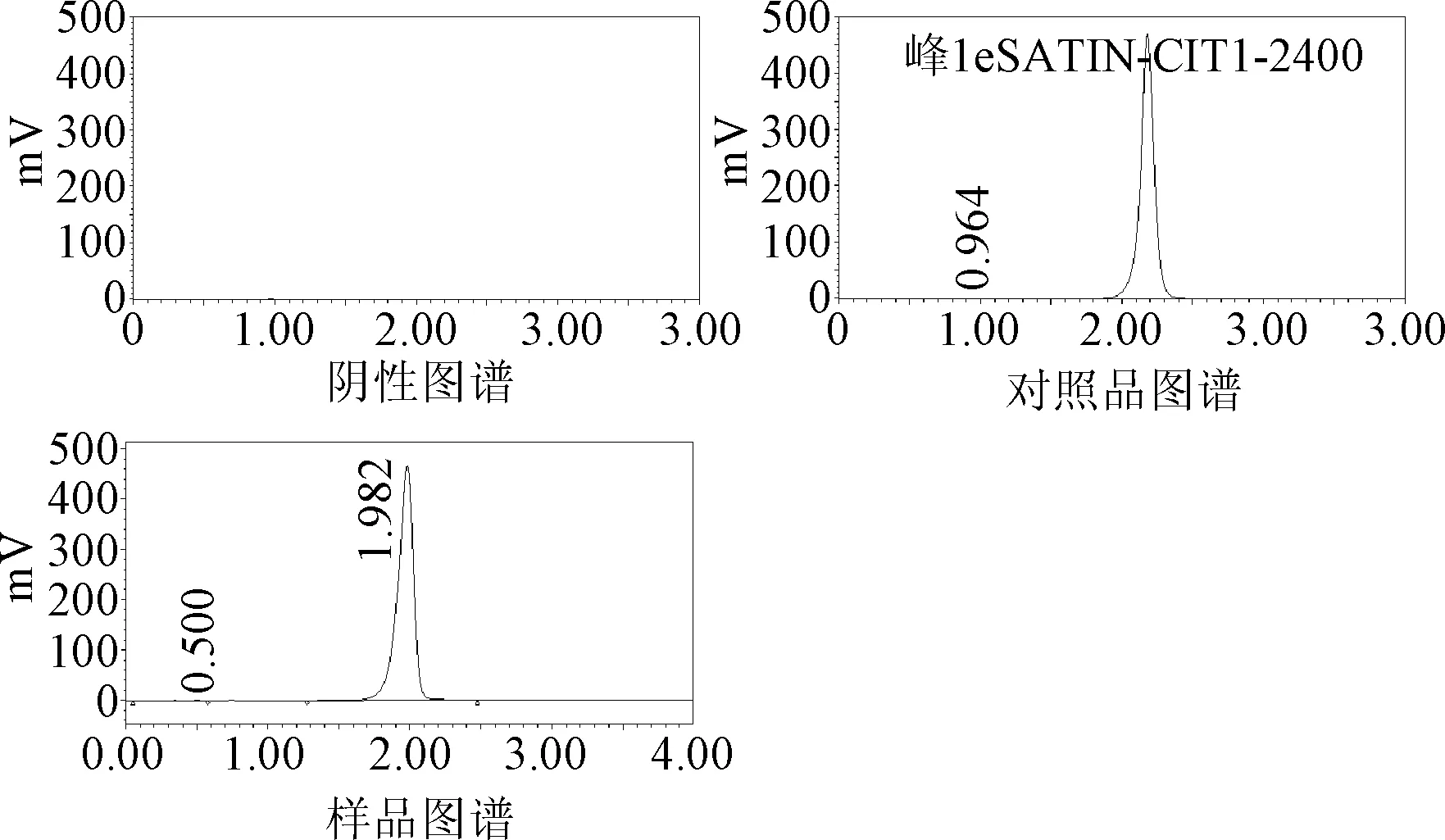
图1 各组分色谱图Fig.1 Chromatogram of each substance
2.1.2 线性:回归方程为: lgy=1.846lgx+2.119,相关系数r=0.9991(n=6),表明盐酸氨基葡萄糖在25~312.5 μg/mL范围内与峰面积线性关系良好。
2.1.3 检测限与精密度:盐酸氨基葡萄糖的检测限为2.5 μg/mL。对照品溶液连续重复6次测得峰面积,RSD为0.6%,仪器精密度良好。同批样品6份计算含量,RSD为0.9%,重现性良好。
2.1.4 加样回收:回收率结果见表1。
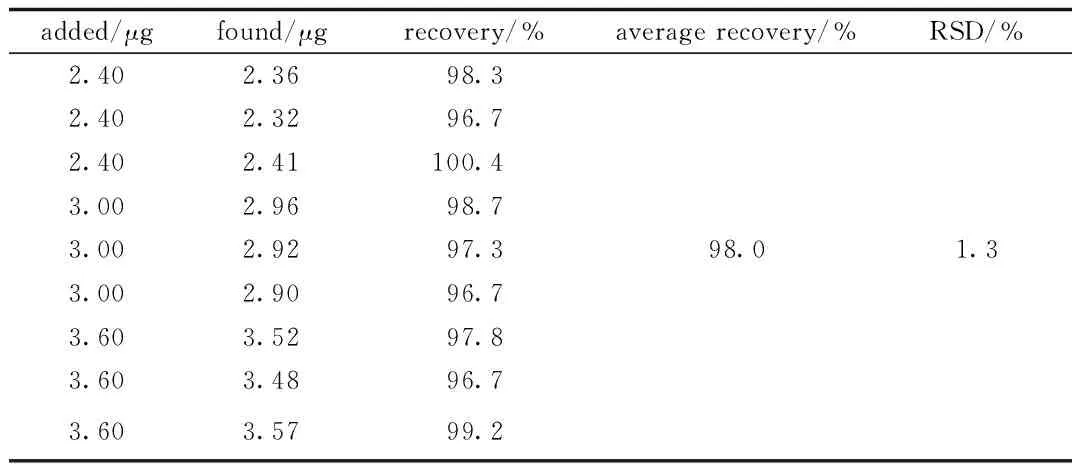
表1 回收率(n=9)
2.1.5 稳定性试验:结果显示样品溶液在0, 3, 6, 9, 12, 18 h进样得到的色谱图,峰面积无明显改变 , RSD为 0.9%,表明样品溶液室温放置18 h内稳定。
2.1.6 样品含量测定:结果见表2。

表2 含量测定(n=5)
2.2 相关物质检测试验结果 酸破坏在11.5 min出现新的杂质峰,但峰型小,碱破坏在0.3和9 min产生了较大的新的杂质峰,加热破坏在5.8 min出现较大的新的杂质峰,强光破坏在0.4 min和9 min产生了较大的新的杂质峰,说明样品对酸较稳定,对碱、加热、强光破坏不稳定,且各破坏条件下,降解产物均能与盐酸氨基葡萄糖峰呈良好分离。
另取样品3批,批号:20150801、20150912、20151103,分别测定,均未检出杂质。
3 讨论
流动相:试用甲醇-水、乙腈-水流动相系统,结果乙腈-水易平衡,且噪音低,比较不同配比的乙腈-水(70:30),峰型良好,故选用乙腈-水(70:30)为流动相。
色谱柱:流动相确定后,比较了离子交换色谱柱、C18谱柱、C8色谱柱。离子交换色谱柱,产生的噪音大,基限漂移。C18和C8柱在相同色谱条件下,C8柱峰型更优,且重复性强,故选用C8柱。
柱温:Rune Slimestad[14]对柱温进行研究,发现温度对葡萄糖测定含量影响较大较,一般控制在20 ℃~30 ℃为宜,故选择柱温25 ℃。
制备样品溶剂:用流动相为溶剂,样品出现2个成分峰,且分离度不理想。通过验证,后1个色谱峰为吲哚美辛峰,尝试调整流动相比例,始终不能分开。考察吲哚美辛的溶解性,水中几乎不溶[15],而盐酸氨基葡萄糖易溶于水,故选用水为溶剂。
[1] 杨淮,王崇益.HPLC-ELSD法测定盐酸氨基葡萄糖胶囊中盐酸氨基葡萄糖的含量[J].淮阳师范学院学报,2015,14(1):48-51.
[2] 薛继杨,王雅萍,张瑶纾.Cu2+络合物紫外法测定盐酸氨基葡萄糖含量[J].中国医药工业杂志,2013,44(4):389-391.
[3] 张德柱,宋愿智,沈登林.盐酸氨基葡萄糖胶囊溶出度测定方法研究[J].西北药学杂志,2014,29(1):54-57.
[4] 王太亮,裴璐,李佳侬.氨糖美辛肠溶胶囊中盐酸氨基葡萄糖含量的测定[J].中国煤炭工业医学杂志,2010,13(3):330-331.
[5] 罗立,但汉雄.氨基柱HPLC法测定盐酸氨基葡萄糖及其制剂的含量[J].中国药师,2016,19(2):387-389.
[6] 廖栩,王淑君,佟岩,等.柱前衍生化法测定盐酸氨基葡萄糖软膏的含量[J].沈阳药科大学学报,2011,28(5):380-383.
[7] 凌林.HPLC-ELSD法测定盐酸赖氨酸葡萄糖注射液的含量[J].药物分析杂志,2007,27(3):454-456.
[8] 吴虹,顾宏霞,王效山.HPLC-ELSD测定氨基葡萄糖含量[J].安徽中医学院学报,2008,27(2):41-43.
[9] 潘柏良,刘霞.HPLC-ELSD法测定盐酸氨基葡萄糖胶囊中主成分的含量[J].中国药房,2013,24(16):1517-1518.
[10] 李海英.HPLC-ELSD检测盐酸氨基葡萄糖中的有关物质[J].华西药学杂志,2008,23(2):229-230.
[11] 訾晨瑞,石秀伟,郝宏山.高效液相色谱-蒸发光散射色谱法法测定盐酸氨基葡萄糖片有关物质的研究[J].天津药学,2011,23(3):18-21.
[12] 靳茂礼,张延峰,汪玉梅.高效液相色谱-蒸发光散射检测器法测定氨基葡萄糖胶囊中有关物质葡萄糖的研究[J].中国药学杂志,2012,47(24):2047-2048.
[13] 张德柱,宋愿智,沈登林.盐酸氨基葡萄糖胶囊有关物质测定方法研究[J].西北药学杂志,2014,29(3):252-255.
[14] Slimestad R, Vagen IM.Thermal stability of giucose and other sugaraldoses in normal phase high performance liquid chromatography[J].J Chromatogr A,2006,1118(2):281-284.
[15] 国家药典编委会 .中华人民共和国药典.二部[M].北京:中国医药科技出版社, 2015:478-479.
(编校:王俨俨)
Determination of glucosamine hydrochloride and related substances in glucosamine and indometacin enteric-coated capsules by HPLC-ELSD
WANG Tai-liang1Δ, WANG Zi-qin2
(1.Tangshan Institute for Food and Drug Control, Tangshan 063000, China; 2.Shanghai Normal University, Shanghai 200235, China)
ObjectiveTo establish an high performance liquid chromatography-evaporative light scattering detector (HPLC-ELSD) method for determination of glucosamine hydrochloride and its related substances in glucosamine and indometacin enteric-coated capsules.MethodsSeparation was carried out on a Inertsil C8Column (250 mm×4.6 mm,5 μm) with a mobile phase composed of acetonitrile-water(70:30) at a flow rate of 1.0 mL/min and the column temperature was 25 ℃.ResultsThe linear ranges of calibration curve was 25-312.5 μg/mL(r=0.9991,n=6).The average recovery was 98.0%, and the relative standard deviation was 1.3%(n=5).The glucosamine hydrochloride was stable with acid destruction, but unstable with alkali, heat and strong light destruction.ConclusionThe method is simple, accurate and reproducible for determination of glucosamine hydrochloride and related substances.
glucosamine hydrochloride; HPLC-ELSD; content determination; related substances
10.3969/j.issn.1005-1678.2016.06.59
唐山市科学技术研究与发展指导计划项目(09130244b)
王太亮,通信作者,男,本科,副主任药师,研究方向:药物分析与检验,E-mail:wangtailiang2006@163.com。
R927
A
猜你喜欢
供水技术(2022年1期)2022-04-19
中国应急管理科学(2021年4期)2021-04-13
昆钢科技(2020年6期)2020-03-29
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
考试周刊(2018年68期)2018-09-17
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
火炸药学报(2014年5期)2014-03-20
郑州大学学报(理学版)(2014年4期)2014-03-01
