
高容量硼酸亲和磁性纳米粒子的制备及其在邻羟基生物分子富集中的应用
2018-09-12 03:11张君才陈佑宁卫引茂
分析化学 2018年9期
张君才 陈佑宁 卫引茂
1(咸阳师范学院化学与化工学院,咸阳712000)2(陕西省现代分离科学重点实验室,西北大学化学与材料科学学院,西安710127)
1 引 言
核苷、糖蛋白等邻羟基类物质在生物体内有重要的生理功能。研究表明,细胞核与细胞质内的糖蛋白参与细胞内的多种应激响应过程,在疾病的发生与治疗中起重要作用[1]。然而,由于糖蛋白在体内的丰度较低且存在复杂的基体干扰,鉴定难度较大,因此,对糖蛋白进行鉴定,必须首先将样品进行分离和富集。目前,已发展了凝集素亲和法[2]、硼酸亲和法[3,4]、肼化学反应法[5]、亲水作用法[6]、β-消除Michael 加成反应法[7]等多种糖蛋白的富集方法。基于磁性硼酸亲和吸附剂的磁性分散固相萃取具有分离简单、萃取时间短的优势,已成为富集糖蛋白/肽的常用方法。Lin[8]和Zhang[9]等分别制备了苯硼酸修饰的磁性吸附剂,并将其用于蛋清中糖蛋白的富集。对于磁性硼酸亲和吸附剂的制备,目前常用的方法是通过共价键将含硼酸的小分子固定于材料表面[8~11],如Zhu等[11]采用分子印迹技术制备的磁性硼酸亲和吸附剂在中性条件下富集了糖蛋白。然而,由于基体表面的吸附层是一种单分子吸附层,所以制备的吸附剂普遍存在吸附容量和吸附效率低的缺点。
研究表明,在粒子表面构建三维聚合物吸附层,能提高吸附剂的吸附容量[12~15]。陈林吉等[16]合成了磁性氮掺杂石墨烯纳米材料, 用于萃取环境水样中的有机氯污染物。表面引发原子转移自由基聚合(SI-ATRP)因其具有活性可控的优点,已成为在材料表面构建三维聚合物吸附层的新方法。通常,SI-ATRP包括固体引发剂的制备和单体原位聚合两步操作反应[17]。对于固体引发剂,目前常采用小分子引发剂对基体表面进行修饰的方法进行制备,该方法虽然简单,但是基体表面引发剂密度低,导致ATRP反应后生成的聚合物刷密度小,限制了吸附容量的进一步提高[12,17]。为提高引发剂的表面接枝密度,本研究采用聚乙烯亚胺(PEI)修饰磁性纳米粒子,再加入引发剂,以增加表面引发剂的密度,通过SI-ATRP将硼酸单体聚合在材料表面,制备了一种新型聚合物刷型硼亲和磁性吸附剂。将此吸附剂用于核苷和糖蛋白的吸附,验证其吸附性能,并将其应用于生物样品分离与富集。
2 实验部分
2.1 仪器与试剂
LC-20A高效液相色谱仪(日本Shimadzu公司),使用InertSustain C18(250 mm×4.6 mm I.D., 5 μm)色谱柱。
FeCl3、柠檬酸钠、无水乙酸钠、盐酸多巴胺(DA)、聚乙烯亚胺(PEI,分子量600)和CuBr(国药集团化学试剂有限公司)。CuBr使用前用10%冰乙酸浸泡过夜,砂芯漏斗过滤,用水反复洗涤至中性,再用甲醇和丙酮洗涤,50℃真空干燥。正硅酸乙酯、2,2'-联吡啶和2-溴异丁酰溴(上海晶纯生化科技有限公司); 3-氨基苯硼酸(APBA,北京百灵威科技有限公司); 乙二醇(天津天力试剂公司); 蛋白Marker(赛默飞世尔科技有限公司); 卵清蛋白(OVA,美国Sigma-Aldrich公司); 溶菌酶(Lys)和牛血清白蛋白(BSA)(西安沃尔森科技有限公司)。
2.2 聚多巴胺包覆Fe3O4磁性纳米粒子(Fe3O4@PDA)的制备
参照文献[18]的方法。采用水热合成法制备F3O4纳米粒子; 将F3O4超声分散于Tris-HCl缓冲液,加入DA,室温下搅拌24 h。磁性粒子用水和乙醇洗涤,干燥后得到Fe3O4@PDA。
2.3 3-丙烯酰胺基苯硼酸(AAPBA)
参照文献[13]的方法制备。在冰浴下,将间氨基苯硼酸加入到 2 mol/L NaOH溶液,搅拌溶解,再滴加丙烯酰氯,室温下反应2 h。溶液调节至pH 1.0,析出白色固体。过滤,滤液用乙酸乙酯萃取,蒸发溶剂。合并所得固体,用水重结晶,得到棕黄色针状晶体,即AAPBA硼酸单体。
2.4 聚乙烯亚胺修饰磁性粒子(Fe3O4@PDA@PEI)的制备
将50 mL 4 mg/mL PEI溶液加入到1.0 g Fe3O4@PDA粒子中,超声分散5 min,机械搅拌5 h。将粒子分别用水、甲醇洗涤3次,得Fe3O4@PDA@PEI粒子,备用。
2.5 固体引发剂的制备
将Fe3O4@PDA@PEI粒子和30 mL四氢呋喃加入到100 mL三颈瓶中,超声分散5 min,再加入0.6 mL三乙胺和0.5 mL 2-溴异丁酰溴,搅拌3 h。粒子分别用四氢呋喃和甲醇洗涤,得固体引发剂,备用。
2.6 硼酸聚合物刷型吸附剂(Fe3O4@PDA@PEI@AAPBA)的制备
将上述固体引发剂分散于20 mL DMF中,加入0.8 g AAPBA和0.2 g 2,2'-联吡啶。混合物经过冷冻-抽真空-解冻循环3次,在N2保护下迅速加入0.1 g CuBr,在95℃反应12 h。用甲醇洗涤后,将粒子浸泡在10% EDTA 溶液中,搅拌2 h,用水和甲醇洗涤,真空干燥,得Fe3O4@PDA@PEI@AAPBA(制备路线见图1)。
2.7 吸附选择性
用竞争吸附法考察吸附剂Fe3O4@PDA@PEI@AAPBA粒子对小分子和生物大分子的选择性。对于小分子物质,用50 mmol/L NH3-NH4Cl(pH 9.0)缓冲液配制5.0 μg/mL的肾上腺素、5-羟色胺、对苯二酚、苯胺、沙丁胺醇和邻苯二酚混合溶液。将5.0 mL混合液和10.0 mg吸附剂加入到离心管中,超声分散,振荡1 h后,磁性分离,弃去上清液。粒子用NH3-NH4Cl缓冲液(pH 9.0)淋洗两次,然后再加入到1.0 mL 乙酸-甲醇(5∶95,V/V)中,振荡30 min,分离洗脱液。洗脱液用HPLC-UV分析检测。
对于生物大分子,实验方法如下:用20 mmol/L 磷酸盐缓冲液(pH 7.0,含0.2 mol/L NaCl)分别配制1.0 mg/mL的BSA、Lys和OVA混合溶液。将20.0 mg粒子加入到4.0 mL蛋白溶液中,超声分散,恒温振荡30 min。磁性分离,弃上清液。粒子用缓冲液淋洗两次,加入1.0 mL 1%乙酸溶液,振荡30 min,磁性分离。洗脱液用十二烷基磺酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)分析。
2.8 吸附容量
用静态吸附法考察吸附剂Fe3O4@PDA@PEI@AAPBA粒子对小分子和生物大分子的吸附容量。
对于小分子,将5.0 mg 吸附剂分散于2.0 mL 1.0 mg/mL邻苯二酚和2.0 mg/mL腺苷的标准溶液 (pH 8.5) 中,恒温振荡30 min,收集上清液,用HPLC-UV测定邻苯二酚和腺苷的浓度,用公式(1)计算吸附容量(Q)。
(1)
其中,Q为吸附容量(mg/g),C0和Ce分别是物质的原始和平衡浓度(mg/mL),V0是溶液体积(mL),m是Fe3O4@PDA@PEI@AAPBA粒子质量(mg)。
对于生物大分子,用0.2 mol/L NaCl+20 mmol/L磷酸盐缓冲液(pH 7.0)分别配制一系列浓度不同的BSA、Lys和OVA溶液。将20.0 mg Fe3O4@PDA@PEI@AAPBA粒子加入到4.0 mL蛋白溶液,超声分散,恒温振荡30 min。收集上清液,用Bradford法[19]测定蛋白浓度,蛋白吸附量用公式(1)计算。
2.9 尿液中核苷的富集与测定
收集健康志愿者尿液,10000 r/min离心15 min。用氨水调节上清液至pH 8.5。在0.9 mL尿液样品中,加入0.1 mL 核苷标准液和5.0 mg Fe3O4@PDA@PEI@AAPBA磁性粒子,超声5 min,再恒温振荡30 min。分离出粒子,用缓冲液(pH 8.5)淋洗3次后,再用100 μL 5%乙酸解吸,用HPLC-UV测定解吸液。以峰面积对核苷的加标浓度作图,制作标准曲线。
2.10 鸡蛋清中糖蛋白的提取
用含有0.2 mol/L NaCl的磷酸盐缓冲液(pH 7.0)将鸡蛋清稀释5倍,以降低粘度,10000 r/min离心5 min,除去不溶物,得到上清液(母液)。将2.0 mL 母液加入到装有20.0 mg Fe3O4@PDA@PEI@AAPBA磁性粒子的离心管中,超声分散5 min,恒温振荡30 min。分离出磁性粒子,将粒子用缓冲液洗涤两次,再用1.0 mL 5% 乙酸溶液解析。母液、上清液与解析液用SDS-PAGE分析。
3 结果与讨论
3.1 Fe3O4@PDA@PEI@AAPBA的制备与表征
在SI-ATRP中,固体表面的引发剂分子一般应为α-位有吸电子诱导效应的卤化物。传统方法是将小分子卤化物通过共价键以单分子层的形式固定于基体表面[12,16]。这种方法引入的引发剂密度一般较低,降低了ATRP反应后聚合物分子刷的密度。为提高聚合物刷的接枝密度, 本研究首先在Fe3O4表面包覆聚多巴胺,然后将支化的PEI修饰到材料表面,再连接上引发剂2-溴异丁酰溴。与传统方法相比,该方法提高了引发剂的密度。然后,用SI-ATRP反应将AAPBA单体原位聚合于粒子表面形成三维吸附层。在吸附过程中,物质可进入聚合物刷内,与侧链上的硼酸进行缩合反应而被吸附。前期研究表明,AAPBA聚合物链的长度随聚合时间的延长而增长,当聚合反应超过8 h,吸附量达到平衡[13],故选取聚合时间为8 h。
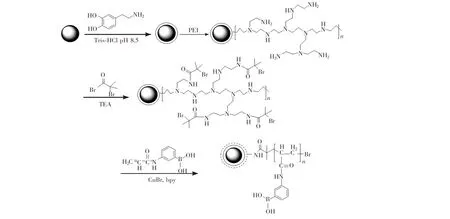
图1 Fe3O4@PDA@PEI@AAPBA粒子的制备路线Fig.1 Illustration of preparation route of Fe3O4@PDA@PEI@AAPBAPEI: polyethylenimine; PDA, polydopamine; bpy, 2,2'-dipyridyl; AAPBA, poly(3-acrylamidophenylboronic acid).
用透射电镜(TEM)和X射线光电子能谱(XPS)对Fe3O4@PDA@PEI@AAPBA进行了表征。与Fe3O4粒子相比,包覆聚多巴胺后,Fe3O4@PDA粒子呈现明显的核-壳式结构,壳层约为20 nm(图2B中的箭头所指红色线条所框区域),说明粒子包覆上了聚多巴胺。经过ATRP反应后,粒子表面的壳层厚度增加到42 nm左右(图2C中箭头所指的红色线条所框区域),说明硼酸聚合物包覆在磁性微球的表面。与Fe3O4相比, Fe3O4@PDA@PEI@AAPBA的XPS图中出现了B元素的峰,测得B元素含量为5.2%。上述结果表明,已成功制备Fe3O4@PDA@PEI@AAPBA。

图2 纳米粒子的TEM图.(A)Fe3O4; (B) Fe3O4@PDA; (C) Fe3O4@PDA@PEI@AAPBAFig.2 Transmission electron microscopy (TEM) image of(A)Fe3O4, (B) Fe3O4@PDA, and (C)Fe3O4@PDA@PEI@AAPBA
3.2 吸附特异性
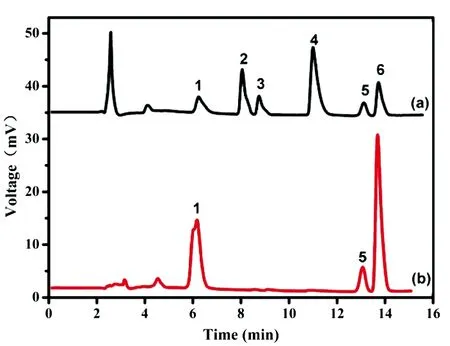
图3 混合物富集前(a)和富集后(b)的色谱图色谱柱:InertSustain C18; 流动相:10 mmol/L NH4Cl 缓冲液-乙腈(85∶15); 流速:1.0 mL/min; 检测波长:254 nm. 色谱峰:1. 肾上腺素; 2. 5-羟色胺; 3. 对苯二酚; 4. 苯胺; 5. 沙丁胺醇; 6. 邻苯二酚Fig.3 Chromatograms of (a) untreated solution and (b) eluent by Fe3O4@PDA@PEI@APPBAColumn: InertSustain C18; Mobile phase: 10 mmol/L NH4Cl-ACN (85∶5); Flow rate: 1.0 mL/min; DetectionV wavelength: 254 nm; Peaks: 1. epinephrine; 2. 5-hydroxytryptamine; 3. hydroquinone; 4. phenylamine; 5. salbutamol; 6. catechol.
在分散固相萃取中,吸附剂的吸附容量以及选择性对目标物的富集效率和分析灵敏度有具有重要作用。文献研究表明,硼酸亲和吸附剂对顺式邻羟基物质的吸附有特异性,但是,多数特异性验证采用的是小分子修饰的吸附剂[8,9,20]。因此,有必要验证三维聚合物吸附层的吸附选择性。首先,采用吸附剂富集6种小分子混合物(肾上腺素、5-羟色胺、对苯二酚、苯胺、沙丁胺醇和邻苯二酚)。从图3可见,只有肾上腺素、沙丁胺醇和邻苯二酚等顺式邻羟基物质被富集,而5-羟色胺、对苯二酚和苯胺3种干扰物质被除去。此外,使用磁性固相萃取对牛血清白蛋白(BSA)、 溶菌酶(Lys)和卵清白蛋白(OVA)的混合液进行萃取。SDS-PAGE电泳结果表明,经过萃取后,解析液中仅存在OVA,说明吸附剂仅对糖蛋白有特异性吸附作用。上述结果表明,三维硼酸聚合物吸附层对邻羟基小分子和生物大分子均具有良好的吸附特异性。
3.3 Fe3O4@PDA@PEI@AAPBA的吸附容量
在已有研究中,常用邻苯二酚和腺苷测定硼酸吸附剂的吸附容量[20~24]。为便于与文献值比较,本研究选择这两个化合物表征 Fe3O4@PDA@PEI@AAPBA的吸附容量。吸附剂对邻苯二酚和腺苷的最大吸附量分别为(151 ± 32) μmol/g和(123 ± 18) μmol/g。与文献报道方法相比(表1),本方法的吸附容量不仅远高于直接键和小分子硼酸配体的方法[20~22],同时还高于聚合物修饰法[15,23,24]。这是因为PEI有效地提高了引发剂位点的密度。 此外,以OVA为模型蛋白,由吸附等温线测得吸附剂的最大吸附量为656.7 mg/g (≈1.5 μmol/g),说明此吸附剂对糖蛋白也有较高的吸附容量。
表1 不同硼酸亲和吸附剂的吸附容量比较
Table 1 Comparison of adsorption capacities of various adsorbents
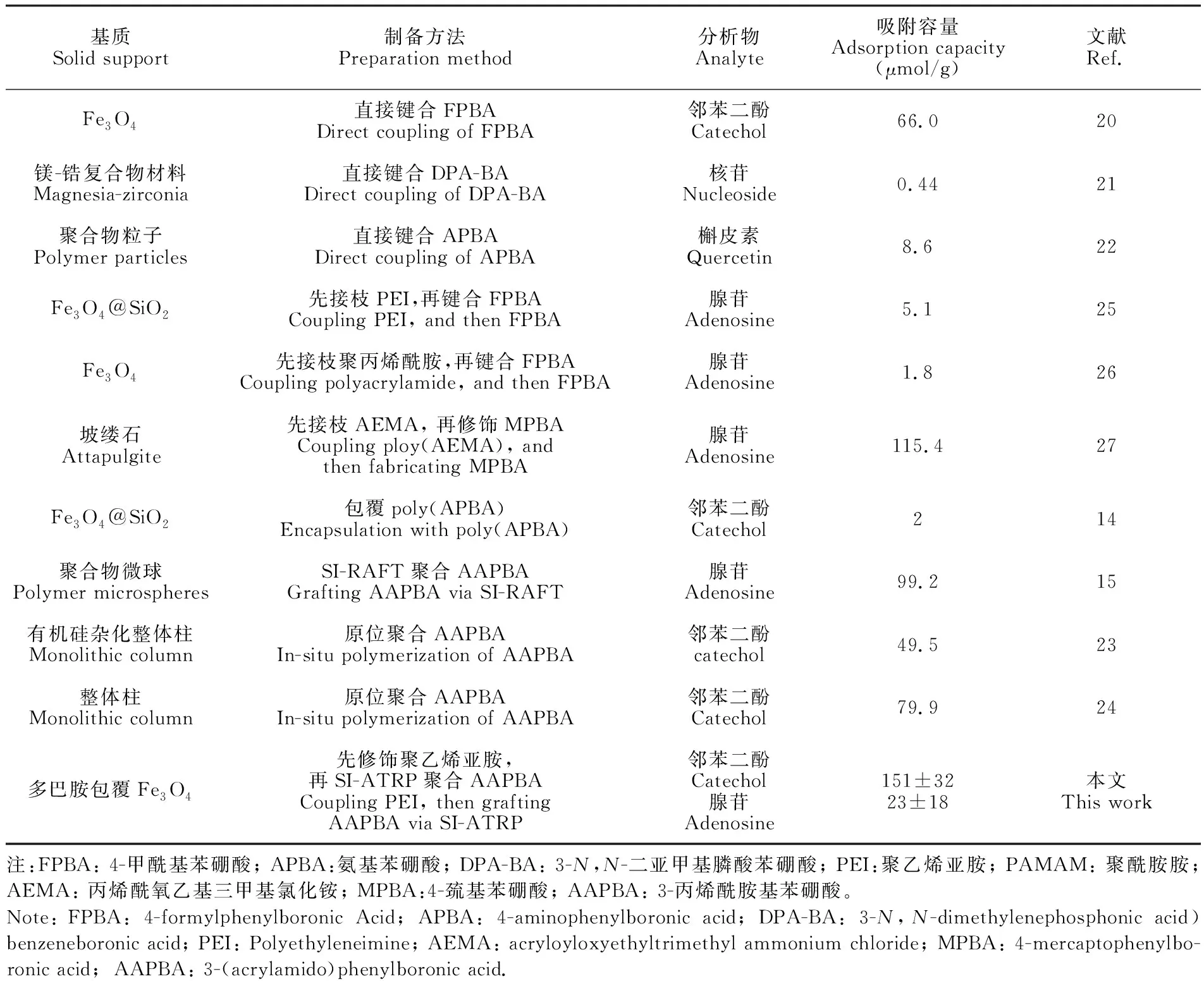
基质Solid support制备方法Preparation method分析物Analyte吸附容量Adsorption capacity(μmol/g)文献Ref.Fe3O4直接键合FPBADirect coupling of FPBA邻苯二酚Catechol66.020镁-锆复合物材料Magnesia-zirconia直接键合DPA-BADirect coupling of DPA-BA核苷Nucleoside0.4421聚合物粒子Polymer particles直接键合APBADirect coupling of APBA槲皮素Quercetin8.622Fe3O4@SiO2先接枝PEI,再键合FPBACoupling PEI, and then FPBA腺苷Adenosine5.125Fe3O4先接枝聚丙烯酰胺,再键合FPBACoupling polyacrylamide, and then FPBA腺苷Adenosine1.826坡缕石Attapulgite先接枝AEMA, 再修饰MPBACoupling ploy(AEMA), andthen fabricating MPBA腺苷Adenosine115.427Fe3O4@SiO2包覆poly(APBA)Encapsulation with poly(APBA)邻苯二酚Catechol214聚合物微球Polymer microspheresSI-RAFT聚合AAPBAGrafting AAPBA via SI-RAFT腺苷Adenosine99.215有机硅杂化整体柱Monolithic column原位聚合AAPBAIn-situ polymerization of AAPBA邻苯二酚catechol49.523整体柱Monolithic column原位聚合AAPBAIn-situ polymerization of AAPBA邻苯二酚Catechol79.924多巴胺包覆Fe3O4先修饰聚乙烯亚胺,再SI-ATRP聚合AAPBACoupling PEI, then graftingAAPBA via SI-ATRP邻苯二酚Catechol腺苷Adenosine151±3223±18本文This work注:FPBA: 4-甲酰基苯硼酸; APBA:氨基苯硼酸; DPA-BA: 3-N,N-二亚甲基膦酸苯硼酸; PEI:聚乙烯亚胺; PAMAM: 聚酰胺胺; AEMA: 丙烯酰氧乙基三甲基氯化铵; MPBA:4-巯基苯硼酸; AAPBA: 3-丙烯酰胺基苯硼酸。Note: FPBA: 4-formylphenylboronic Acid; APBA: 4-aminophenylboronic acid; DPA-BA: 3-N,N-dimethylenephosphonic acid) benzeneboronic acid; PEI: Polyethyleneimine; AEMA: acryloyloxyethyltrimethyl ammonium chloride; MPBA: 4-mercaptophenylbo-ronic acid; AAPBA: 3-(acrylamido)phenylboronic acid.
3.4 聚合刷型硼酸亲和磁性吸附剂在实际样品中的应用
3.4.1尿液中核苷的富集与测定图4是用Fe3O4@PDA@PEI@AAPBA粒子对加标尿液进行富集前后的色谱图。由图4可见,富集前的尿液样品中含有大量的杂质峰 (谱线c),严重干扰目标物的测定; 经过富集之后,许多干扰峰消失,同时4种核苷的色谱峰面积明显增大(谱线b)。 说明Fe3O4@PDA@PEI@AAPBA微球能选择性富集核苷,同时能够有效消除复杂生物样品中的基质干扰。
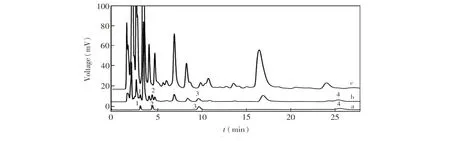
图4 尿液样品的色谱图. (a)核苷标准品; (b) 萃取的加标尿液; (c)加标0.2 μg/mL核苷的尿液。色谱柱:InertSustain C18; 流动相:25 mmol/L KH2PO4(pH 4.52)-甲醇,20 min内甲醇比例从5%线性变化到60%。流速:1 mL/min,检测波长:260 nm。色谱峰1:胞苷,2:尿苷,3:鸟苷,4:腺苷。Fig.4 Chromatograms of standard solution of nucleosides (a), eluate of the spiked urines after extraction (b) and the urines spiked with 0.2 μg/mL of 4 nucleosides (c). Column: InertSustain C18; mobile phase: 25 mmol/L KH2PO4(pH 4.52)-methanol, methanol volume ratio linearly changes from 5% to 60%. Flow rate, 1 mL/min. Detection wavelength, 260 nm. Peaks: 1. cytidine; 2. uridine; 3. vernine; 4. Adenosine.
采用标准加入法,在尿液中加入一系列浓度不同的4种核苷,通过峰面积对加标浓度作图,获得4种核苷的标准曲线。尿苷的标准曲线方程为y=79268x+13442 (R2=0.989); 鸟苷为y=85431x+30881 (R2=0.996); 胞苷为y=63294x+40864 (R2=0.985); 腺苷为y=69742x+33109 (R2=0.991)。用外推法求得尿液中尿苷、鸟苷、胞苷和腺苷的浓度分别为0.17、0.36、0.65和0.48 μg/mL,该数值处于健康人体核苷的浓度范围之内[28]。此外,由表2可见,高、中、低加标水平尿液经过富集后的回收率为83.8%~108.7%,且RSD < 15 %,说明本方法具有良好的准确度和精密度,可满足生物样品定量分析的要求[29]。
表2 4种核苷的富集回收率
Table 2 Recoveries and precisions for determination of 4 kinds of nucleosides
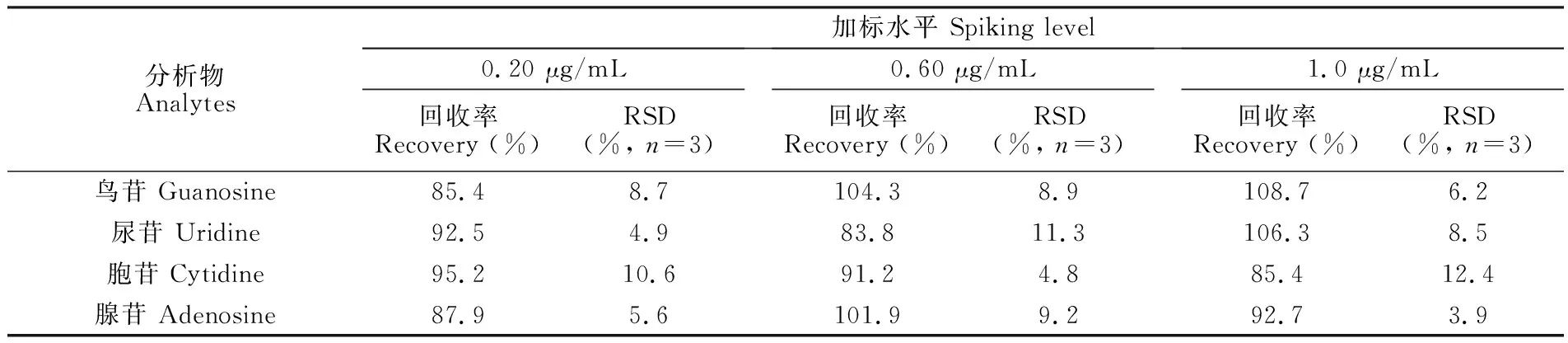
分析物Analytes加标水平 Spiking level0.20 μg/mL回收率Recovery (%)RSD(%, n=3)0.60 μg/mL回收率Recovery (%)RSD(%, n=3)1.0 μg/mL回收率Recovery (%)RSD(%, n=3)鸟苷 Guanosine85.48.7104.38.9108.76.2尿苷 Uridine92.54.983.811.3106.38.5胞苷 Cytidine95.210.691.24.885.412.4腺苷 Adenosine87.95.6101.99.292.73.9
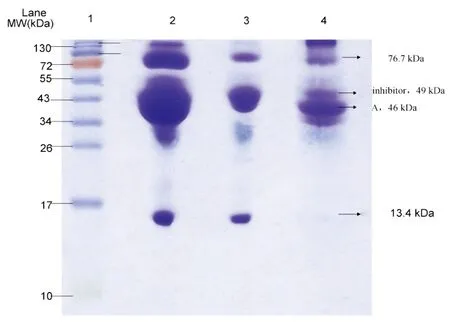
图5 蛋清样品的电泳图。条带1:Marker; 条带2:未处理的蛋清样品; 条带3:上清液; 条带4:洗脱液。Fig.5 Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of egg white samples. Lane 1, protein marker; Lane 2, untreated egg white; Lane 3, supernatant; Llane 4, eluate
3.4.2鸡蛋清中糖蛋白的富集鸡蛋清中主要包含卵转铁蛋白 (12%,pI 6.1)、卵清蛋白(54%,pI 4.5)、卵粘蛋白(1.5%~3.5%,pI 4.5~5.0)、溶菌酶 (3.5%,pI 10.7)、卵球蛋白 G2(4.0%,pI 5.5)、类卵粘蛋白(11%,pI 4.1) 和卵球蛋白 G3(4.0%,pI 4.8)。 其中,类卵粘蛋白、卵清蛋白、转铁蛋白和卵球蛋白属于糖蛋白,而溶菌酶则属于非糖蛋白。蛋清样品经过磁性萃取,用SDS-PAGE电泳检测上清液、洗脱液和蛋清样品(图5)。 与蛋清原液相比(条带2),上清液电泳图中糖蛋白颜色相对变浅(条带3),而溶菌酶条带颜色几乎保持不变,这是由于吸附剂吸附了蛋清中的糖蛋白,使上清液中糖蛋白浓度减少,而对非糖蛋白基本上没有吸附,从而使溶菌酶留在上清液。从电泳条带4可见,洗脱液主要含有3种糖蛋白,而没有溶菌酶。此结果说明吸附剂在复杂生物样品中可特异性地捕获糖蛋白,同时能够去除非糖蛋白。
4 结 论
采用聚乙烯亚胺修饰磁性纳米粒子,再加入引发剂,通过SI-ATRP法在粒子表面原位聚合硼酸聚合物链,制备了一种聚合物刷型硼酸亲和磁性吸附剂。由于在纳米粒子表面修饰了高密度引发剂,提高了ATRP反应后粒子表面的硼酸聚合物链密度,故此吸附剂对小分子和糖蛋白有较高的吸附容量。同时,此吸附剂对邻羟基小分子和生物大分子具有吸附特异性。将此吸附剂用于人体尿液和蛋清的分离富集,能够选择性吸附邻羟基生物分子和糖蛋白,排除干扰分子,说明此吸附剂在生物样品分离富集领域具有良好的应用潜能。
猜你喜欢
化学分析计量(2021年6期)2021-06-22
闽南师范大学学报(自然科学版)(2020年1期)2020-06-05
生物技术通报(2019年9期)2019-09-18
中成药(2018年7期)2018-08-04
中成药(2018年1期)2018-02-02
中成药(2017年5期)2017-06-13
中成药(2017年5期)2017-06-13
现代检验医学杂志(2016年1期)2016-11-12
医学研究杂志(2015年3期)2015-06-10
中国当代医药(2015年9期)2015-03-01
