
以消化道症状为首发表现的家族性淀粉样多发神经病一家系报道并文献复习
2019-11-29 03:03张丽环张晓燕王贺波
疑难病杂志 2019年11期
张丽环,张晓燕,王贺波
家族性淀粉样多发神经病(familial amyloid polyneuropathy,FAP)属罕见的常染色体显性遗传性疾病,通常是由于蛋白质基因突变导致水溶性蛋白形成类淀粉纤维,在细胞外沉积而致病。相关的致病蛋白有转甲状腺素蛋白(transthyretin,TTR)、载脂蛋白 A-1和凝溶胶蛋白(gelsolin),其中以TTR型最为常见[1]。TTR基因的点突变是导致 TTR型FAP的主要原因,目前已在TTR基因上发现近一百多个相关的突变点位与缺损,最常见的是Val30Met(18号染色体长臂第30个氨基酸由原本的缬氨酸突变为甲硫氨酸)突变,在全球范围内约占1/2,且分布于不同种族间[2-3]。疾病早期多以下肢感觉异常为首发症状,逐渐累及运动神经,伴有自主神经病变,最后死于恶病质。尽管感觉和运动表现是主要症状,但首发临床表现可以是自主神经病变症状[4]。现对一个以消化道症状为首发表现的FAP家系进行分析总结,并进行相关文献复习。
1 临床资料
患者,女,39岁。主因“胃胀、饮食不佳5年,四肢无力3个月,加重20 d”于2018年1月31日入院。患者5年前出现胃胀、饮食不佳,伴恶心,偶有呕吐,伴腹泻,每日3~4次,于当地医院就诊行胃镜检查示慢性非萎缩性胃炎,给予中药、护胃等药物治疗后自觉症状稍有改善。近2年来患者仍有胃胀、饮食不佳、腹泻症状。3个月前出现活动后肢体无力,休息后缓解。后症状逐渐加重,日常体力活动受影响,步行1 km即感肢体无力;入院前20 d患者肢体无力症状明显加重,持筷子费力,不能系扣子,不能正常蹲起,常伴头晕发作,尤以起立时显著,偶伴心慌,曾晕厥1次。患者自发病以来精神可,饮食、睡眠差,体质量较1年前轻约7 kg。既往体健。入院查体:T 36℃,P 79次/min,R 19次/min,BP 119/88 mmHg。发育正常,重度营养不良。心肺腹检查未见明显异常。专科查体:神清语利,高级皮质功能正常,颅神经检查未见异常。双上肢肌力Ⅳ+级,双手拇间肌萎缩,双下肢远端肌力Ⅳ-级,近端肌力Ⅳ+级,双下肢腱反射减弱,浅感觉减弱,深感觉无明显异常,双侧Hoffmann征阴性,双侧Babinski、Chaddock 征未引出,颈无抵抗,Kernig 征阴性。
辅助检查:甲状腺功能六项、凝血四项、电解质、女性肿瘤全项、乙肝五项+丙抗、粪便分析、血同型半胱氨酸、血清铁代谢、抗核抗体、抗核抗体谱未见明显异常。生化全项:血清总蛋白62.4 g/L,白蛋白39.45 g/L。尿液分析:酮体(±),上皮细胞47.7/μl。血常规:白细胞2.09×109/L,中性粒细胞0.99×109/L,红细胞计数3.27×1012/L,血红蛋白98 g/L。维生素B12>2 000 pg/ml。骨髓穿刺涂片:骨髓增生较低下(约20%),粒红比例减低,粒系各阶段细胞可见,以中幼及以下阶段细胞为主;红系以中晚幼及以下红细胞为主;巨核细胞多见,分叶核为主;淋巴细胞、浆细胞散在分布。24 h动态血压平均值94/64 mmHg,白昼血压平均值83/53 mmHg,夜间血压平均值94/64 mmHg。心电图:V1、V2导联呈QS型,肢体导联QRS波低电压,V5、V6导联电压小于1.0 mV。24 h动态心电图:存在短阵室速。心脏超声心动图示:心肌弥漫性病变(心肌淀粉样变),二、三尖瓣少量反流,左心室舒张功能减低,少量心包积液(见图1)。腹部超声示:膀胱、胆囊沉积物。头颅MR检查未见明显异常。胸部CT示:右肺下叶外基底段及双肺下叶后基底段微小结节,双肺下叶后基底段小条索,少量心包积液。神经电生理检查:被检测肌肉呈神经元性损害,四肢周围神经损害(运动、感觉纤维均受累);基因检测:患者TTR基因c.148G>T(p.V50L)杂合变异,未在受检者母亲外周血中检出(见图2)。其余相关家属拒绝抽血未行基因检测。最终诊断家族性淀粉样多发神经病。给予注射用硫辛酸、甲钴胺注射液营养神经,右佐匹克隆改善睡眠,地榆升白片提升白细胞,莫沙比利促进消化,胺碘酮抗心律失常等药物治疗。出院时患者胃胀及肢体无力等症状略有缓解,神经系统查体较前无明显变化。
家系调查:一个家系4代共5例患者(男4例,女1例),分别为先证者(Ⅳ5)及其大伯(Ⅲ1)、父亲(Ⅲ3)、表叔(Ⅲ8)及二哥(Ⅳ3),除先证者外均已故。患者家系图见图3。
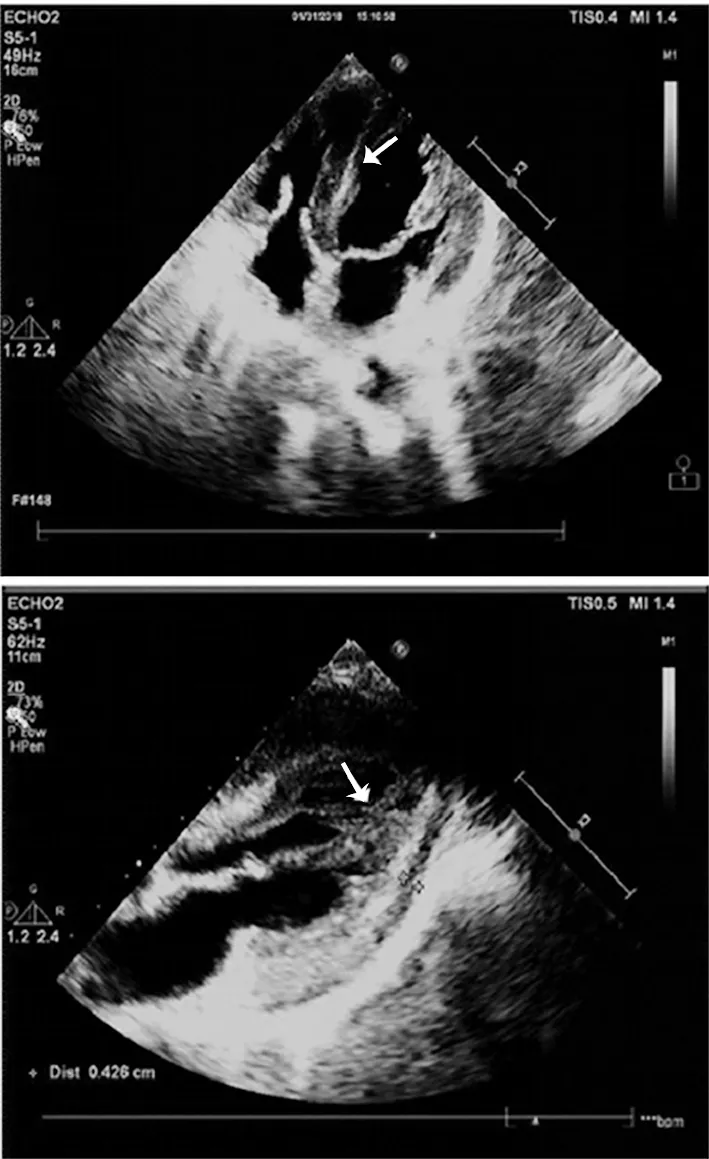
图1 心脏超声心动图示心尖四腔心切面及胸骨旁长轴切面心肌淀粉样变性(箭头显示室壁增厚,回声减低,)
2 讨 论
TTR型FAP是一种罕见疾病,最早于1952年由葡萄牙北部的Andrade报道。据统计此病在葡萄牙及瑞典、日本、巴西相对高发,其发病率在1/10 000 ~1/1 000,其中95%~99%为TTR Val30Met突变,这种突变占家族性淀粉样多发神经病世界移植登记处(FAPWTR)报道的TTR突变的85%[5]。欧洲的发病率估计为每年0.003/万,美国发病率估计为1/10万。
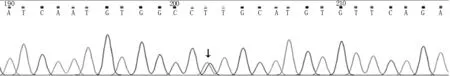
图2 先证者基因检测结果:TTR基因测序图发现c.148G>T(p.V50L)杂合变异(如箭头所示)
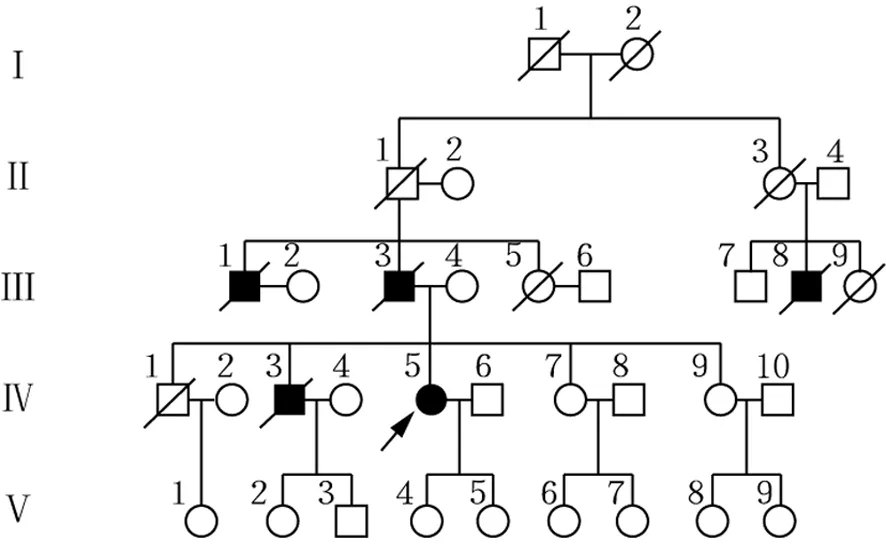
注:□正常男性;○正常女性;/已逝者;■男性患者;●女性患者;→先证者
图3 FAP患者家系图谱
而我国东北、福建、香港、台湾及澳门等地亦有FAP家系报道,其中李延峰等[6]在2001年首次从临床、病理等方面报道了我国FAP一家系病例。刘晶瑶等[7]在2012年详细阐述了此病临床表现及基因表型。由TTR基因c.148G>T突变所致FAP病例十分少见,仅Suhr等[8]曾报道过1例66岁芬兰病例。而杨硕等[1]在2017年也报道了一家系以足部疼痛为首发症状的早发型FAP,填补了我国对TTR基因c.148G>T突变致FAP研究的空缺。
FAP可根据地理分布、致病蛋白及临床表现的不同分为5型(见表1)。FAPⅠ型(日本、葡萄牙型)是世界最常见的FAP类型,1978年Costa最早证实葡萄牙FAP患者的淀粉样沉积物为TTR基因点突变所致,其主要临床特征是缓慢进展的感觉运动性神经病和自主神经功能障碍。通常是从下肢末端(脚趾不适)的感觉神经开始出现异常如皮肤感觉异常,温觉和痛觉异常较早出现[9]。疾病的中期,感觉缺失呈手套—袜套样分布并累及运动神经出现肌无力及肌萎缩。自主神经功能障碍症状突出,主要损害心脏循环系统、泌尿生殖系统和胃肠道系统,导致心脏传导衰竭、直立性低血压、阳痿、恶病质、发作性恶心呕吐、胃轻瘫、腹泻、便秘或二者交替、排尿困难、性功能障碍及泌汗异常等。后期患者卧床不起或依靠轮椅生存,表现为恶病质,平均存活10年左右[10]。中枢神经损害表现为精神行为异常,脑神经损害及锥体束征。眼睛受累表现包括玻璃体混浊伴进行性视力丧失、小梁阻塞所致慢性开角型青光眼和扇形瞳孔[11]。也可累及肾脏发生渐进性肾衰竭。据报道,超过2/3的TTR基因突变者伴有心肌病[12],典型超声心动图表现为心室壁增厚伴颗粒状或斑点状心肌,左心室体积小,舒张受限[13]。在心脏淀粉样变性的突变位点中,Val122Ile在非洲裔美国人群中发病率显著增高,其中3.0%~3.9% 的突变基因为杂合子[14]。本例家系临床表现和FAP-I型特别类似,患者的大伯、父亲、表叔、二哥都是以消化道症状为首发,逐渐出现感觉运动功能症状,同时伴有自主神经功能障碍,最后因过于消瘦死于恶病质。而患者除以上症状外,出现心功能不全症状,同时心电图及动态心电图有病理改变,心脏超声显示心肌淀粉样变性。神经肌电图呈神经元性损害。基因分析发现c.148G>T(p.V50L)杂合变异,可确定诊断FAPⅠ型。
FAP的早期诊断是至关重要的,但由于表型异质性、症状出现较晚,诊断延迟和误诊仍很常见。在意大利最近的一项研究中,此病误诊率高达32%,从最初的症状到确诊约3年,约20%误诊为慢性炎性脱髓鞘多神经病,其次是腰骶神经根病、腰椎管狭窄、副蛋白血症性神经病和系统性轻链淀粉样变性(AL淀粉样变性)[15]。对于有FAP家族史的患者,如果接受了基因检测,组织活检并不是必须的[11]。本例家系先证者以消化道症状为首发表现,后逐渐出现多发性感觉运动神经病,心脏超声检查发现弥漫性心肌损害,提示淀粉样物质沉积,结合其多发性感觉运动神经病、明确家族史,考虑TTR-FAP可能,遂仅行TTR基因检测,结果提示c.148G>T(p.V50L)杂合变异而得以明确诊断。
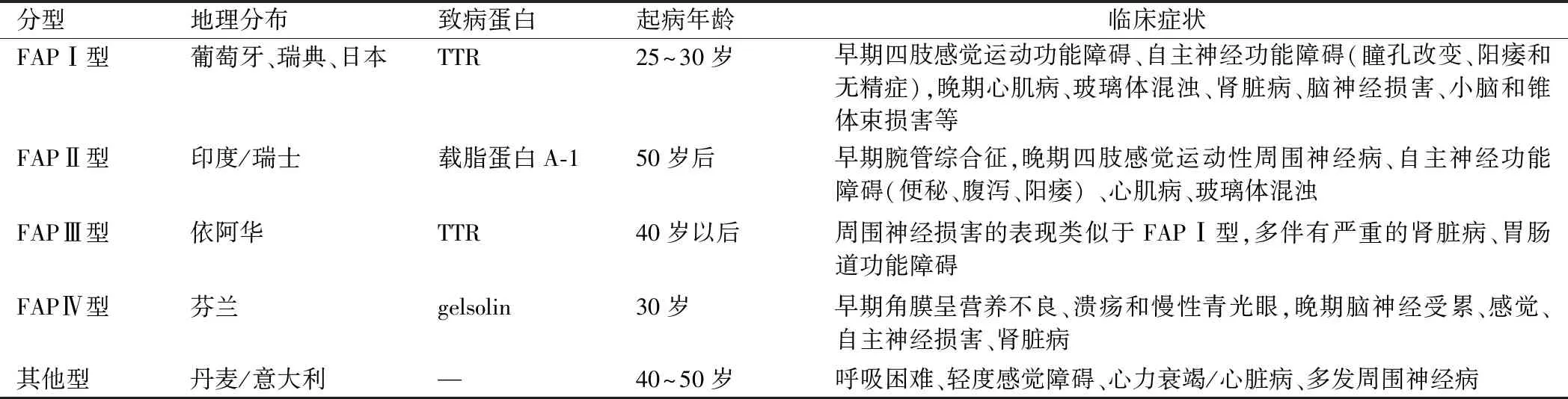
表1 家族性淀粉样周围神经病分型
但因先证者曾祖、祖父一代去世早,时间久远,家人无法回忆当时情况,不能确定此2代人患病情况,但此家系自第Ⅲ代开始表现为明确的常染色体显性遗传,符合TTR-FAP临床特征。
TTR是一种同源四聚体蛋白,主要产生于肝脏,少量产生于脉络膜丛和视网膜细胞[16],因此,对于早发患者可行肝移植(LT)治疗,LT是TTR-FAP的第一个治疗方案,其15年生存率接近80%[17-18],而未行肝移植的患者10年生存率仅为 56.1%[19]。目前一种转甲状腺素蛋白稳定剂药物(氯苯唑酸),通过与甲状腺素结合位点结合稳定突变的TTR四聚体,防止其分解成单体,减少淀粉样蛋白形成,可能改善神经系统和心脏情况的进展[20-21]。二氟尼柳是20世纪70年代开发的非甾体抗炎药(NSAID),是体外形成TTR淀粉样蛋白纤维的强抑制剂,它可以防止突变TTR四聚体的分离、错折叠和错组装。Berk等[22]的一项多中心研究表明,二氟尼柳可抑制多神经病进展,提高TTR-FAP患者不同疾病阶段的生活质量。目前的一种基因沉默疗法[使用小干扰RNA (siRNA)或反义寡核苷酸ASO],通过强烈降低突变体和野生型TTR血浆水平,可抑制TTR蛋白的产生[23]。目前正在研究淀粉纤维的靶向药物抗血清淀粉样蛋白P(SAP)制剂或抗TTR抗体,其目标在于清除组织中已经存在的淀粉样蛋白沉积,以减少淀粉样蛋白负荷,最终恢复器官功能[24]。采用脂质纳米粒子技术的ALN-TTR01和ALN-TTR02是一种系统性RNA干扰疗法,旨在抑制TTR蛋白的表达,防止淀粉样蛋白的形成[25]。对于所有TTR-FAP突变携带者,无论有无症状,都应定期进行神经、心脏、肾脏和眼部检查。神经检查包括感觉、运动和反射评估,肌电图、体位血压和心率变异性检测。心脏筛查包括进行心电图和超声心动图,测量BNP或氨基末端脑钠肽前体(NT-pro-BNP)。除此之外,对症治疗也很重要,针对神经痛可以应用卡马西平、加巴喷丁、普瑞巴林等药物;针对心脏受累后出现传导阻滞可以安装起搏器;针对体位性低血压等症状可以加用药物及调整生活方式控制改善症状[26]。
综上,TTR-FAP是一种罕见的、TTR基因突变所致的、表现为多发性感觉运动神经病的全身性疾病,部分患者可能以消化道症状等自主神经损害为首发症状。对于具有家族史的、以多发性感觉运动神经病为主要表现的患者,如超声提示心脏淀粉样物质沉积,应考虑TTR-FAP诊断可能,行TTR基因检测可明确。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国典型病例大全(2022年10期)2022-05-10
保健医苑(2022年4期)2022-05-05
广西医科大学学报(2021年11期)2021-12-20
基层中医药(2018年11期)2019-01-31
吉林林业科技(2018年6期)2018-11-21
中国循环杂志(2018年8期)2018-09-03
科普童话·神秘大侦探(2017年4期)2017-04-06
现代临床医学(2016年6期)2016-02-20
听力学及言语疾病杂志(2015年5期)2015-12-24
