
线粒体tRNA亮氨酸基因第3251位点A到G点突变致线粒体肌病伴高乳酸血症1家系报告
2021-12-23 10:21郭雯琪任可欣王迎新
中风与神经疾病杂志 2021年11期
郭雯琪, 任可欣, 曹 华, 王迎新
线粒体tRNA亮氨酸基因(mitochondrial tRNALeu(UUR) gene (UUR,R=A or G),MT-TL1,OMIM:590050)突变能够导致线粒体疾病。MT-TL1包含线粒体DNA(mitochondrial DNA,mtDNA)第3230-3304碱基对。目前已经发现的MT-TL1的致病性点突变有:3242 G>A,3243 A>G,3249 G>A,3250 T>C,3251 A>G,3256 C>T,3260 A>G,3271 T>C,3274 A>C,3290 T>C,3303 C>T等[1~12]。MT-TL1最常见的突变为3243 A>G点突变[1~12],特征性地表现为线粒体脑肌病伴高乳酸血症和卒中样发作综合征(mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes,MELAS)。而MT-TL1 3251 A>G点突变的患者相当少见。自1993年至今,世界范围内仅公开发表了3个家系的病例报告[1~3];2020年复旦大学儿童医院报告的对21例中国线粒体肌病患儿进行基因测序,发现了1例MT-TL1 3251 A>G点突变患儿[4]。因此,由于病例稀少,目前对MT-TL1 3251 A>G点突变所致的线粒体疾病的致病性及临床异质性认识还不充分,需要不断积累病例。现报告我们发现的1个MT-TL1 3251 A>G点突变的中国家系。
1 临床资料
先证者,女,20岁,汉族。因“反复发作性胸闷、全身无力8 y,再发5 d,加重伴尿潴留1 d”于2013年11月21日入住大连医科大学附属第一医院急诊科,经会诊后转入神经内科。患者自8y前12岁时开始出现反复发作性胸闷、全身无力,严重时不能行走,自诉发作时在外院诊治时发现多种酶学指标增高(具体不详),给予对症治疗后好转,未确诊。类似严重发作有3次。12岁时,她因此病辍学了。近2 m来患者做家务较劳累,入院前5 d再次出现胸闷、全身无力、心率增快,双下肢近端肌肉疼痛,可以平地行走几步,抬手及上台阶无力,恶心,入院前3~4 d能正常饮水,未进食物。入院前1 d上述症状加重,伴尿潴留,在当地医院急诊留置导尿后转来我院急诊。既往史:否认其他疾病。个人史:足月顺产,自幼较同龄人运动能力差,智能发育正常,身高较矮153cm(父母身高均为160cm)。为父母的独生女儿。家族史:亲属中无肌无力患者。母亲自30多岁起患有糖尿病,50岁时出现肾功衰竭、双目失明。父亲体健。入院时查体:体温36.0 ℃,脉搏142次/min,呼吸24次/min,血压125/57 mmHg。神清,语明。精神、智能正常。双眼上睑轻度下垂,双侧面肌无力,颈肌和躯干肌无力,不能抬头,不能在床上翻身。四肢肌肉容积减少,双下肢肌张力减低,四肢肌力2级,四肢腱反射对称性减低。头部MRI正常。心电图:HR 142 bpm,窦性心动过速。血液化验显示她患有乳酸酸中毒:动脉血气分析:pH 7.004,PCO222.6 mmHg,PO287.0 mmHg,HCO3-act 5.5 mmol/L,HCO3-std 8.0 mmol/L,剩余碱-24.4 mmol/L↓;血浆乳酸浓度14.2 mmol/L↑(参考值:0.7~2.1 mmol/L);血清CK 5820 IU/L↑(参考值:90~135 IU/L),CK-MB 767.2 μg/L↑(参考值:0~5 μg /L)。血常规:WBC 18.50×109/L↑,N 79.01%↑,RBC 3.77×1012/L,Hb 116.0 g/L,PLT 168×109/L。ALT 138 IU/L↑(参考值:13~69 IU/L),AST 435 IU/L↑(参考值:15~46 IU/L)。甲状腺功能、肾功能、血糖、电解质均正常。心脏彩超正常。腹部彩超正常。肌电图示肌源性损害:运动单位动作单位(motor unit action potential,MUAP)呈现波幅降低,时限缩短。神经传导速度正常。右侧股外侧肌活检显示肌束间和肌束膜内纤维脂肪组织增生。HE染色显示大量稍蓝染嗜盐基破碎肌纤维,少部分肌纤维内含有大小不等的数个小泡,可见部分萎缩肌纤维和再生肌纤维,部分肌纤维核内移。改良GOMORI-TR染色(modified Gomori trichrome,MGT)可见部分破碎红纤维(ragged-red fibers,RRF)。细胞色素C氧化酶(cytochrome C oxidase,COX)染色显示散在分布的COX(-)肌纤维;COX/琥珀酸脱氢酶(succinate dehydrogenase,SDH)染色可见COX(-)肌纤维SDH(+)。抗线粒体内膜(mitochondrial inner membrane(3H2248),MIM,(SANTA CRUZ:sc-71589))抗体免疫组织化学染色(immunohistochemistry,IHC)显示受累肌纤维肌膜下MIM染色强(+)的线粒体积聚。抗肌球蛋白重链新生型[myosin heavy chain(neonatal),MHC-n,(Leica,NCL-MHCn)]抗体IHC显示几个MHC-n(+)肌纤维(见图1)。油红O染色显示部分肌纤维含有过多脂质。PAS染色没有检测到糖原沉积。外周血mtDNA测序显示MT-TL1 3251 A>G点突变,外周血中G突变峰约28%(见图2)。进一步对她的父母和亲属进行mtDNA测序,显示她的母亲同样有MT-TL1 3251 A>G点突变,其外周血中该突变负荷为23.3%(见图2,家系图见图3)。她的母亲自30多岁时即患有糖尿病,血浆乳酸水平轻度升高3.6 mmol/L(参考值:0.7~2.1 mmol/L)。综上,该青年女患者被诊断为母系遗传的MT-TL1 3251 A>G点突变所致的线粒体肌病伴高乳酸血症。给予高糖低脂饮食、吸氧、静脉滴注甲泼尼龙注射液40 mg/d×6 d,静点复合辅酶注射液、肌肉注射维生素B1与维生素B12注射液2w;长期口服三磷酸腺苷(adenosine triphosphate,ATP)片、丁苯酞软胶囊、维生素B1片、维生素B2片、维生素B6片、辅酶Q10和腺苷钴胺片。入院1w后她的病情明显减轻,双下肢肌肉疼痛缓解,能够独立行走、自行上楼梯了。住院12 d后,她的症状缓解了,出院前复查心电图:HR 81 bpm,窦性心律不齐。再次进行血液化验结果为CK 162 IU/L↑(参考值:90~135 U/L),CK-MB 28.45 μg/L↑(参考值:0~5 μg/L)。血浆乳酸浓度 5.97 mmol/L↑(参考值:0.7~2.1 mmol/L)。经随访8 y,她可以独立行走和做家务,再无上述急性发作。她的母亲在53岁时因尿毒症去世。

A:HE染色×100。左下角箭头显示脂肪组织增生;其余箭头指向肌内膜下稍蓝染嗜盐基肌纤维;B:COX染色×100。箭头指向散在分布的COX(-)肌纤维;C:MGT染色×200。箭头指向RRF;D:MHC-n染色×100。箭头指向MHC-n (+)肌纤维;E:SDH染色×100。箭头指向SDH深染蓝纤维;F:COX/SDH染色×100。箭头指向COX(-) SDH (+)肌纤维;G:抗线粒体内膜抗体染色×100。箭头指向散在分布的染色强(+)肌纤维图1 先证者右股外侧肌活检染色图片

A:先证者外周血m. 3251 A>G,G峰约占28%;B:先证者母亲外周血m. 3251 A>G,G峰约占23.3%图2 mtDNA基因测序结果
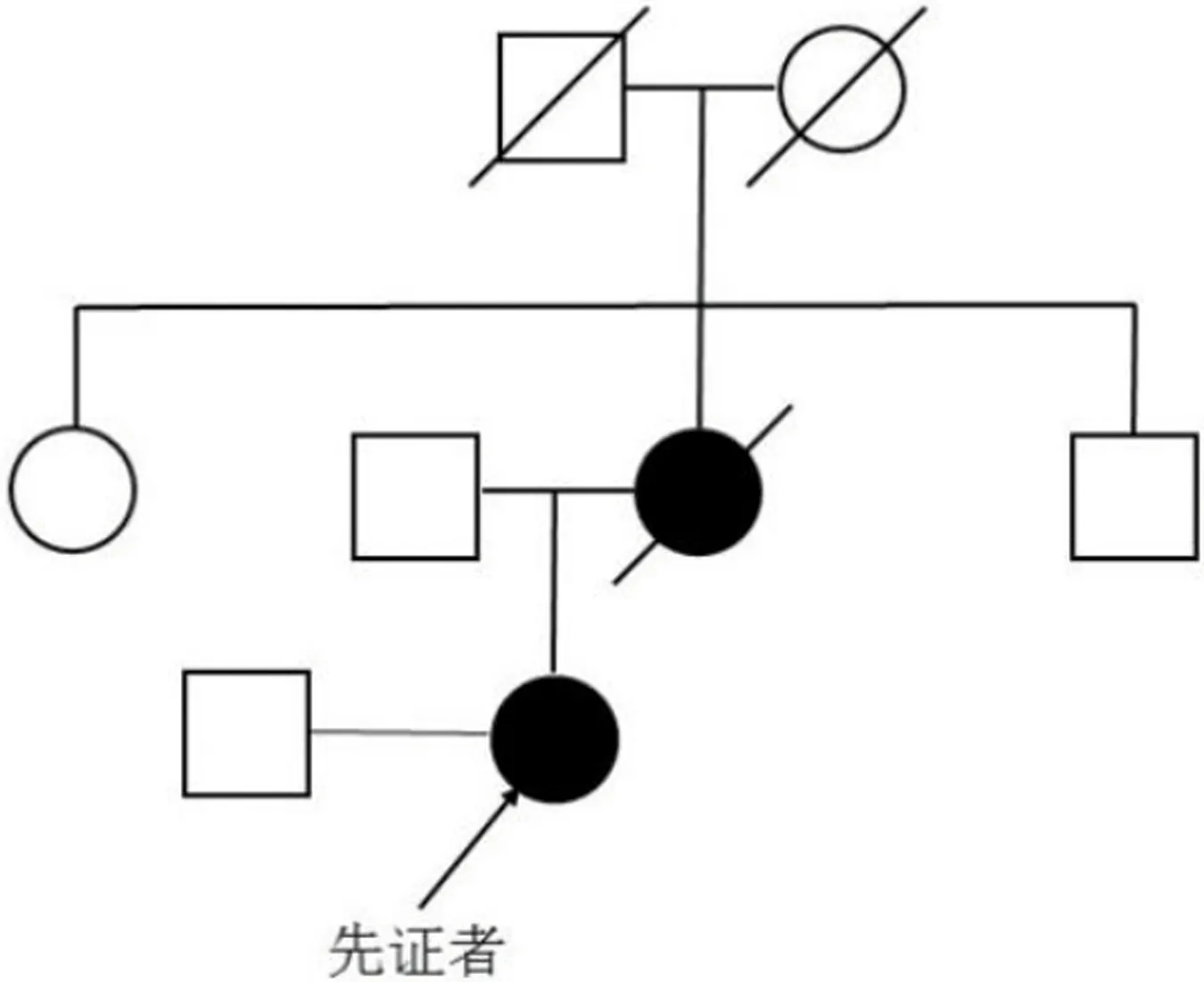
图3 家系图
2 讨 论
青年女患,自幼表现为运动不耐受,病情因疲劳、饥饿加重时会表现为心动过速、肌无力加重,血清CK明显升高。否认肌肉病家族史。此次由劳累与饥饿诱发出现严重肌肉无力、心动过速、恶心、乳酸酸中毒。血清CK和乳酸水平显著升高。经过休息、吸氧、补充ATP、复合辅酶和甲泼尼龙注射液治疗,症状明显改善,运动能力稳步提高。肌肉活检发现散在分布的COX(-)肌纤维[COX代表呼吸链复合体IV,由mtDNA和细胞核DNA(nucleus DNA,nDNA)基因共同编码[9,11,12]];COX/SDH染色发现呈马赛克分布的COX(-)SDH(+)肌纤维(SDH代表呼吸链复合体Ⅱ,完全由nDNA编码[9,11,12]);COX(-)纤维的马赛克分布提示mtDNA突变(肌细胞间的异质性变化)。MIM IHC显示受累肌纤维肌膜下线粒体积聚。以上结果支持该线粒体肌病源于mtDNA突变。基于以上,mtDNA基因测序显示MT-TL1 3251位点A>G点突变,外周血中G突变峰约28%。她母亲也有该点突变,外周血中该突变负荷为23.3%。综上,该青年女患者被诊断为母系遗传的MT-TL1 3251 A>G点突变所致线粒体肌病、心动过速、恶心和乳酸酸中毒;她的母亲表现为线粒体糖尿病。这个中国家系为继续丰富MT-TL1 3251 A>G点突变的致病性及临床异质性提供了进一步的临床、生化学、肌肉活检以及遗传学证据。
在哺乳类和鸟类,腺嘌呤(adenine,A)正常地保守地位于3251位点碱基上,在MT-TL1基因的二氢尿嘧啶核苷环上,它位于与3243碱基相对的位点上[10]。迄今为止,只有4篇文献报告了MT-TL1 3251 A>G点突变。1993年,Sweeney 等[1]首先报告了一个线粒体肌病伴有青年猝死的与MT-TL1 3251 A>G点突变相关的英国家系。1996年,Houshmand等[2]报告了瑞典的一个MT-TL1 3251 A>G点突变的散发病例,这个女孩的父母和2个兄弟体健,患有间断发作的胃肠道征象:轻度用力时腹泻、恶心、疲劳、心动过速。13岁时,她全身肌无力和消瘦,尤其是肩胛带和骨盆带肌。14岁时,患者出现心肺衰竭、高碳酸血症,心肺复苏后死亡。2013年,Mancuso等[3]报告了1例意大利的成年男患,有进行性眼外肌麻痹和双上肢肌无力,突发呼吸衰竭,肌肉活检发现RRF和COX(-)肌纤维,伴散在炎细胞浸润和坏死肌纤维,mtDNA分析揭示了MT-TL1 3251 A>G点突变,除呼吸机辅助呼吸外,试验性给予免疫球蛋白注射液和卡尼汀治疗,临床效果极好。2020年,复旦大学儿童医院的Hu等[4]发表了1组有24个病例的中国儿童线粒体肌病患者的临床研究,有21例患儿完成了基因检测,有1例儿童mtDNA分析发现MT-TL1 3251 A>G点突变,列表显示患儿在10岁时起病,表现为肌无力、运动不耐受、胃肠道受累、心肌受累、呼吸衰竭,16岁时死亡,没有其他的家系资料。
MT-TL1不同位点突变可以导致不同的临床表现,确切机制目前尚不清楚。即使是同一点突变,如最常见的MT-TL1 3243 A>G点突变,也表现为一系列表型,推测可能与不同的突变的mtDNA比例和年龄相关。
相信随着相关病例的逐渐积累和对该基因功能的深入研究,在不远的将来,科学家们将会进一步阐明MT-TL1 基因不同位点突变导致不同的临床表型的确切机制。
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
中国临床医学影像杂志(2022年5期)2022-07-26
医学美学美容(2021年21期)2021-11-20
临床误诊误治(2021年3期)2021-03-30
国际放射医学核医学杂志(2021年10期)2021-02-28
中华养生保健(2021年18期)2021-02-13
新疆大学学报(自然科学版)(中英文)(2020年2期)2020-07-25
医学新知(2019年4期)2020-01-02
蚌埠医学院学报(2016年5期)2016-12-16
安徽医科大学学报(2015年9期)2015-12-16
