
1例早发脑型肝豆状核变性家系ATP7B基因突变和临床表型分析
2021-12-23 10:52钱南南王久香杨文明
中风与神经疾病杂志 2021年11期
钱南南, 王久香, 裴 培, 杨 悦, 杨文明
肝豆状核变性(Wilson’s Disease,WD,MIM 277900)是一种常染色体隐性遗传的以铜代谢紊乱为特征的代谢性疾病,主要表现为肝功能异常、神经或精神损害、角膜可见K-F环(Kayser-Fleischer ring)甚至肾损伤。临床上,该病主要分为肝型(肝功能损伤)、脑型(神经精神异常)和混合型(二者都异常)。患者一般在5~35岁发病,儿童以肝型为主,脑型患者少见[1,2]。目前发现WD是由ATP7B基因突变所导致,我们对1例中国汉族早发型脑型WD家系进行ATP7B变异鉴定,以明确其遗传学病因,为该家系的疾病诊断提供遗传学证据,对家系其他成员的再发风险进行指导。
1 对象与方法
1.1 对象 先证者,女,13岁。2017年7月份渐出现口角流涎、言语不清,写字慢、字体歪斜。就诊于当地卫生所考虑“胃炎”,予口服药治疗未见好转(具体用药不详)。发病16 m开始出现左手不自主抖动,记忆力明显减退。患者双眼角膜K-F环阳性,脑部MRI显示双侧基底节区、黒质对称性异常低信号(见图1),24 h尿铜 621.7 μg/d,ALT、AST、GGT和AKP正常,CER 0.093 g/L(参考值0.2~0.6 g/L),考虑“肝豆状核变性”,予排铜、保脑等综合治疗后口角流涎及左手不自主抖动较前好转。患者4岁时患过病毒性脑炎。

表1 ATP7B外显子及其侧翼区PCR扩增引物序列
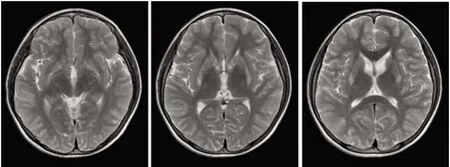
先证者脑部MRI显示双侧基底节区、黒质对称性异常低信号图1 先证者头部MRI
本研究遵循《赫尔辛基宣言》,经中国合肥安徽中医药大学第一附属医院伦理委员会批准,本研究参与者均签署知情同意书。
1.2 方法
1.2.1 基因组DNA提取 EDTA抗凝管采集先证者及家系其他成员的外周血3 ml(见图2A),全血基因组DNA提取试剂盒(北京天根生化科技有限公司)提取基因组DNA,并用分光光度计(Eppendorf,德国)检测其浓度和质量。
1.2.2 基因测序 利用Primer Premier 5 软件设计扩增包含ATP7B基因外显子及其外显子内含子连接区引物,由武汉擎科生物有限公司合成(见表1)。PCR体系(50 μl):5X PrimeSTAR缓冲液(含Mg2+)10 μl (Takara)、基因组DNA 100 ng、上下游引物各0.2 μmol/L、dNTP Mixture 0.2 mmol/L(Takara),PrimeSTAR DNA polymerase 0.5U(Takara),用H2O补足至50 μl体系。PCR反应条件:98 ℃预变性5 min;98 ℃ 变性10 s;55 ℃~60 ℃退火5 s,72 ℃延伸1 min,共35个循环,最后72 ℃延伸10 min,4 ℃保存。PCR产物送武汉擎科生物有限公司用ABI PRISM 3100测序仪测序。测序结果与NCBI数据库中的ATP7B基因序列进行比对(见图2B-2E)。
2 结 果
ATP7B基因测序结果:外显子直接测序结果显示,先证者ATP7B基因存在c.2333G>T和c.2975C>T复合杂合错义变异(见图2C),分别导致ATP7B第778位精氨酸变成亮氨酸,第992位脯氨酸变成亮氨酸。经检索HGMD数据库、dbSNP、千人基因组等数据库证实,c.2333G>T和c.2975C>T均为已知WD致病位点。先先证者母亲携带c.2333G>T(p.Arg778Leu)突变(见图2B),先证者父亲携带c.2975C>T(p.Pro992Leu)突变(见图2C)。先证者的弟弟携带c.2333G>T(p.Arg778Leu)突变(见图2E)。
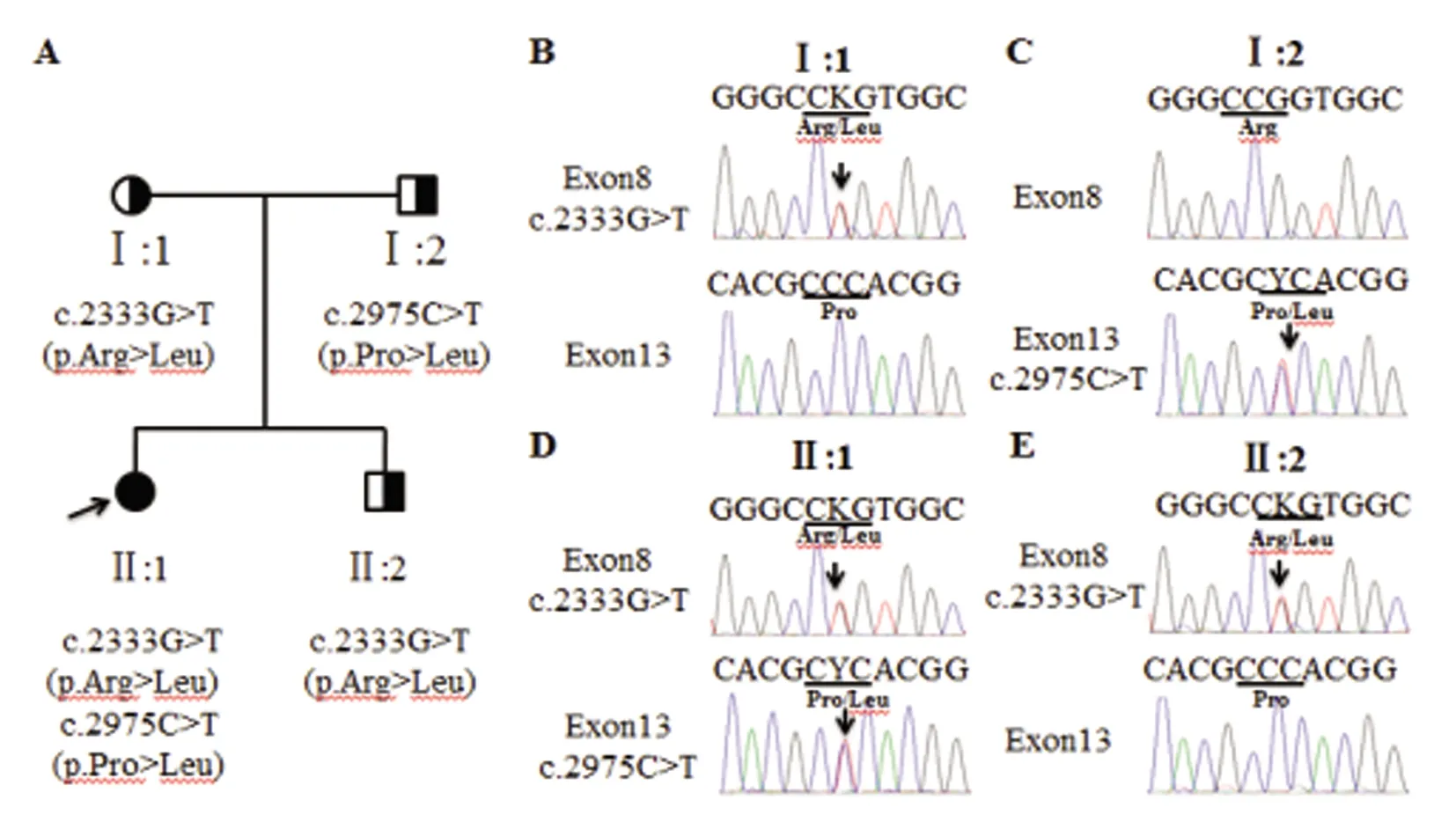
A:先证者家系图,箭头指示先证者;B~E:ATP7B测序结果(正向),箭头指示变异位点图2 先证者家系及测序结果图
3 讨 论
WD是由于体内铜离子异常累积导致的一种罕见隐形遗传病。ATP7B是目前已知的WD致病基因,其定位于13q14.3,编码含有1465氨基酸残基的跨膜蛋白。铜离子的代谢障碍导致其在脑部、肝脏、眼底甚至肾脏累积,导致患者出现肝硬化、神经精神紊乱、角膜K-F环甚至肾损伤。因此早诊断、早治疗可减少患者肝、脑等组织的损伤,延缓病情进展,明显提高WD患者的生存质量,最终回归家庭和社会。患者自出生即存在铜代谢障碍,一般3岁以后才开始出现症状,临床表现各异且大部分为非特异性,因此早期诊断仍较为困难[3]。本文先证者发病初期口角流涎、言语不清,曾考虑胃炎,外院治疗后未见好转。发病4 m后,流涎、言语不清加重,开始出现左手不自主抖动。
目前ATP7B基因已经发现有600多个WD致病突变位点。H1069Q是目前中欧最常见的WD突变位点,在波兰、罗马尼亚,奥地利等中欧国家的等位基因频率为30%~60%[4]。R778L是日本、韩国、中国等亚洲患者的热点变异位点[5]。R778L等位基因在韩国WD患者的携带率约为 37.9%、日本约为27%,中国约28%~44%[4,6,7]。c.2975C>T(p.Pro992Leu)突变位于ATP7B第8外显子,蛋白的跨膜结构域。2013年,有学者报道了1例9岁的中国WD男童,携带与本文先证者相同的突变位点ATP7B c.2333G>T和c.2975C>T[8]。该患者神经精神正常,本文中的先证者有明显的神经精神症状;角膜K-F环阴性,本文患者阳性;肝功能指标ALT(137 IU/L)、AST(106 IU/L)明显升高,本文先证者ALT(16 U/L,参考值5~35 U/L)、AST(22 U/L,参考值5~35 U/L)。有多位学者尝试寻找患者基因型和发病年龄、性别、临床表型的关系,但是没有明确的结论[9~11]。最新研究显示ATP7B DNA甲基化可能与患者临床表型相关[12]。此外先证者4岁时曾患有病毒性脑炎,是否与患者的早期发病相关亦未可知。
先证者的母亲携带ATP7B c.2333G>T突变,父亲携带c.2975C>T突变。先证者从双亲遗传到ATP7B c.2333G>T和c.2975C>T符合杂合突变,结合患者临床症状、生化检查及体征,诊断WD明确。通过对先证者的弟弟ATP7B 基因21个外显子和外显子内含子衔接区的测序发现其只携带c.2333G>T突变,提示其弟弟的发病可能性不大,可密切随访观察。
我们报告了一例早发型脑型WD家系,明确该家系WD发病的遗传学基础是ATP7B c.2333G>T和c.2975C>T,并对先证者的弟弟进行突变检测,分析其患病的可能性。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
实用肝脏病杂志(2022年2期)2022-03-21
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
中国生殖健康(2020年4期)2021-01-18
诊断学(理论与实践)(2020年1期)2020-04-28
水产科学(2020年2期)2020-03-20
森林工程(2018年1期)2018-05-14
