
消渴脉康颗粒质量标准研究
2014-05-03 06:15何绍斌林以慈罗钧锦
中国药业 2014年5期
何 洋 ,何绍斌 ,金 文 ,林以慈 ,罗钧锦 ,石 钺
(1.中国医学科学院药用植物研究所,北京 100193; 2.广州市悠好福医药科技有限公司,广东 广州 510610)
消渴脉康颗粒是由黄芪、枸杞子、桑寄生、赤芍、丹参、地黄、牛膝、延胡索等组方,由医学名著《医学心悟》中治疗消渴病的黄芪汤化裁而来,具有健脾益肾、活血通络之功效,用于治疗糖尿病周围神经病变。经过多年临床应用证实,其具有较好的临床疗效。为有效控制药品的质量,确保该制剂的临床疗效,本试验对组方中各药材建立了定性鉴别方法,对方中主药赤芍的有效成分芍药苷建立了高效液相色谱(HPLC)定量测定方法。试验结果显示,所建方法简便易行、灵敏准确、专属性强,可用于消渴脉康颗粒的质量控制。
1 仪器与试药
CAMAG薄层色谱扫描仪;Waters 1525型高效液相色谱仪(Waters 1525系统,Waters 2487检测器,Mellennium32色谱工作站);TU-1800型紫外分光光度计;KQ-250DB型数控超声波清洗器(220 V,250 W,40 kHz);Mettler AB135-S 型十万分之一电子天平。枸杞子对照药材(批号为121072-201109)、齐墩果酸对照品(批号为 110709-201206)、原儿茶醛对照品(批号为110810-201007)、槲皮素对照品(批号为 100081-200907)、延胡索乙素对照品(批号为110726-201213)、黄芪甲苷对照品(批号为110781-200613)、芍药苷对照品(批号为110736-201337),均购自中国食品药品检定研究院。消渴脉康颗粒(广州市悠好福医药科技有限公司,批号为 20130404,20130405,20130406)。薄层层析用硅胶G和硅胶 GF254,购自青岛海洋化工厂。甲醇为色谱纯,水为纯净水,其他试剂均为分析纯。
2 方法与结果
2.1 薄层色谱(TLC)定性鉴别
2.1.1 牛膝
取本品 10 g,研细,加甲醇 - 水(8 ∶2)100 mL,硫酸 1 mL,加热回流1 h,滤过,滤液浓缩至20 mL,用石油醚(60~90℃)提取2次,每次20 mL,提取液蒸干,残渣加乙醇2 mL使溶解,作为供试品溶液。另取齐墩果酸对照品,加乙醇制成每1 mL各含0.5 mg的溶液,作为对照品溶液。再取缺牛膝的消渴脉康颗粒同法制备阴性对照品溶液。照 2010年版《中国药典(一部)》附录Ⅵ B中TLC法试验,吸取上述3种溶液各 20 μL,分别点于同一硅胶 G薄层板上,以氯仿 -甲醇(40∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照品溶液色谱则无此斑点(见图1 A)。
2.1.2 丹参
取2.1.1项下石油醚提取后的水溶液,用乙醚提取2次,每次20 mL,提取液蒸干,残渣加乙醇2 mL使溶解,作为供试品溶液。另取原儿茶醛对照品,加乙醇制成每1 mL各含1 mg的溶液,作为对照品溶液。再取缺丹参的消渴脉康颗粒同法制备成阴性对照品溶液。照2010年版《中国药典(一部)》附录ⅥB中TLC法试验,吸取上述3种溶液各10 μL,分别点于同一硅胶 GF254薄层板上,以氯仿 - 丙酮 - 甲酸(7.9 ∶1.1 ∶1)为展开剂,展开,取出,晾干,置紫外光灯(254 nm)下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照品溶液色谱则无此斑点(见图1 B)。
2.1.3 枸杞子
取2.1.2项下乙醚提取后的水溶液,用乙酸乙酯提取2次,每次20 mL,提取液蒸干,残渣加乙醇2 mL使溶解,作为供试品溶液。另取枸杞子对照药材1 g,同法制成对照药材溶液。再取缺枸杞子的消渴脉康颗粒同法制成阴性对照品溶液。照2010年版《中国药典(一部)》附录ⅥB中TLC法试验,吸取上述3种溶液各10 μL,分别点于同一硅胶G薄层板上,以醋酸乙酯 -氯仿-甲酸(2.5 ∶2 ∶1)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色的荧光斑点,阴性对照品溶液色谱则无此斑点(见图1 C)。
2.1.4 桑寄生
取本品 20 g,研细,加甲醇 - 水(8 ∶2)100 mL,硫酸 1 mL,加热回流1 h,滤过,滤液浓缩至20 mL,用石油醚(60~90℃)提取2次,每次20 mL,弃去,水溶液继续乙醚提取,提取2次,每次30 mL,合并提取液,蒸干,残渣加乙醇2 mL使溶解,加到聚酰胺1 g中,拌匀,干燥后,加至聚酰胺柱(2 g),先用30 mL水洗脱,弃去,继续用30 mL甲醇洗脱,收集洗脱液,蒸干,残渣用甲醇2 mL溶解,作为供试品溶液。另取槲皮素对照品,加乙醇制成每1 mL含1 mg的溶液,作为对照品溶液。再取缺桑寄生的消渴脉康颗粒同法制成阴性对照品溶液。照2010年版《中国药典(一部)》附录ⅥB中TLC法试验,吸取上述3种溶液各20 μL,分别点于同一用2%氢氧化钠溶液制备的硅胶G薄层板上,以甲苯(水饱和)-甲酸乙酯 -甲酸(5∶3∶1)为展开剂,展开,取出,晾干,喷以 5%三氯化铝乙醇溶液显色,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照品溶液色谱则无此斑点(见图1 D)。
2.1.5 延胡索
取本品20 g,研细,加水100 mL,加热回流4 h,滤过,滤液通过AB-8型大孔吸附树脂柱(内径2.0 cm,长10 cm),以水100 mL洗脱,弃去水液。再用10%乙醇50 mL洗脱,弃去10%乙醇洗脱液,续用95%乙醇80 mL洗脱,收集洗脱液70 mL,浓缩至10 mL,作为供试品溶液。另取延胡索乙素对照品,加无水乙醇制成每1 mL含1 mg的溶液,作为对照品溶液。再取缺延胡索的消渴脉康颗粒同法制成阴性对照品溶液。照2010年版《中国药典(一部)》附录ⅥB中TLC法试验,吸取上述3种溶液各20 μL,分别点于同一硅胶G薄层板上,以石油醚(60~90℃)-醋酸乙酯(1∶1)为展开剂,展开,取出,晾干,置碘蒸气中熏数秒钟后,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照品溶液色谱则无此斑点(见图1 E)。
2.1.6 黄芪
吸取 2.1.5项下供试品溶液 5 mL,蒸干,残渣加30 mL水溶解,用水饱和的正丁醇提取2次,每次20 mL,合并正丁醇液;用氨试液洗涤2次,每次20 mL,弃去氨液,正丁醇液蒸干,残渣加甲醇1 mL使溶解,加到硅胶1 g中,拌匀,干燥后,加至2 g硅胶柱,用30 mL甲醇洗脱,收集洗脱液,蒸干,残渣用甲醇2 mL溶解,作为供试品溶液。另取黄芪甲苷对照品,加甲醇制成每1 mL含1 mg的溶液,作为对照品溶液。再取缺黄芪的消渴脉康颗粒同法制备成阴性对照品溶液。照2010年版《中国药典(一部)》附录Ⅵ B中 TLC法试验,吸取上述3种溶液各2 μL,分别点于同一硅胶 G薄层板上,以氯仿-甲醇-水(13∶7∶2)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品溶液色谱中,在与对照品溶液色谱相应位置上,日光下显相同的棕褐色斑点,紫外光灯(365 nm)下显相同的橙黄色荧光斑点,阴性对照品溶液色谱则无此斑点(见图1 F)。
2.2 芍药苷HPLC法定量测定
2.2.1 色谱条件
色谱柱:安捷伦公司 Zorbax SB-C18柱(250 mm×4.6 mm,5 μm);流动相:甲醇 -0.05 mol/L 磷酸二氢钾(29 ∶71);流速:1.0 mL /min;柱温:30 ℃ ;检测波长:230 nm;进样量:10 μL。
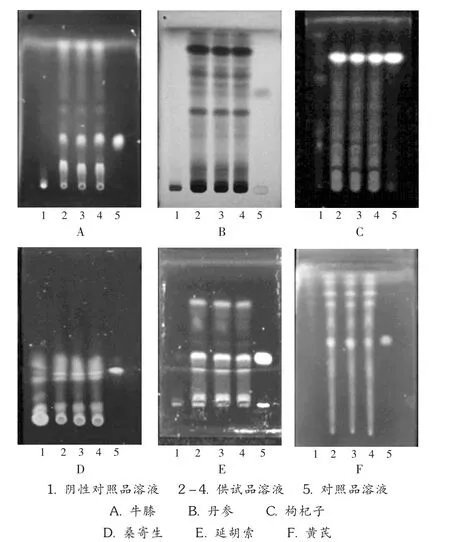
图1 薄层色谱图
2.2.2 溶液制备
精密称取经五氧化二磷减压干燥器中干燥36 h的芍药苷对照品适量,加甲醇制成每1 mL含0.117 2 mg的溶液,即得对照品溶液。取消渴脉康颗粒(批号为20130404)约1 g,精密称定,置具塞锥形瓶中,精密加入50%乙醇25 mL,称定质量,超声处理40 min,放冷,再称定质量,用50%乙醇补足减失的质量,摇匀,滤过,取续滤液,即得供试品溶液。根据消渴脉康颗粒处方,取除赤芍外的其他处方量药材,按质量标准制备方法制成阴性对照品。按供试品溶液制备方法操作,即得阴性对照品溶液。
2.2.3 方法学考察
专属性试验:分别取 2.2项下 3种溶液各 10 μL,注入高效液相色谱仪,记录色谱图。理论板数以芍药苷峰计大于3 000;供试品溶液色谱中芍药苷与相邻峰为基线分离,分离度大于1.5;阴性对照品溶液色谱中无芍药苷的色谱峰,说明测定无干扰。见图2。
线性关系考察:分别精密量取对照品溶液 3.0,5.0,7.0,10.0,13.0,15.0 μL 注入高效液相色谱仪中,按拟订色谱条件测定,以对照品进样量(X)为横坐标、相应峰面积积分值(Y)为纵坐标,绘制标准曲线,得回归方程 Y=130 584 X-137 510(r= 0.999 5)。结果表明,芍药苷进样量在 0.266 4 ~ 1.332 0 μg范围内与峰面积呈良好线性关系。
精密度试验:精密吸取同一对照品溶液,按拟订色谱条件测定5次。结果的 RSD为0.98%(n=5),表明仪器精密度较高,可靠性强。
稳定性试验:取同一供试品溶液,按拟订色谱条件分别于0,2,4,6,8 h 时各测定 1 次。结果的 RSD 为 0.28%(n=5),表明供试品溶液在8 h内稳定性良好。
重复性试验:取同一批样品(批号为20130404)5份,按拟订色谱条件测定芍药苷的含量。结果的 RSD为2.37%(n=5),表明方法重现性较好。
加样回收试验:取批号为20130404的样品(芍药苷含量0.325 3%)研细,取约 0.5 g,精密称取 6份,分别加入对照品溶液10 mL,按供试品溶液的制备方法操作,精密吸取供试品溶液10 μL,按拟订色谱条件测定,计算回收率。结果见表1。

图2 高效液相色谱图
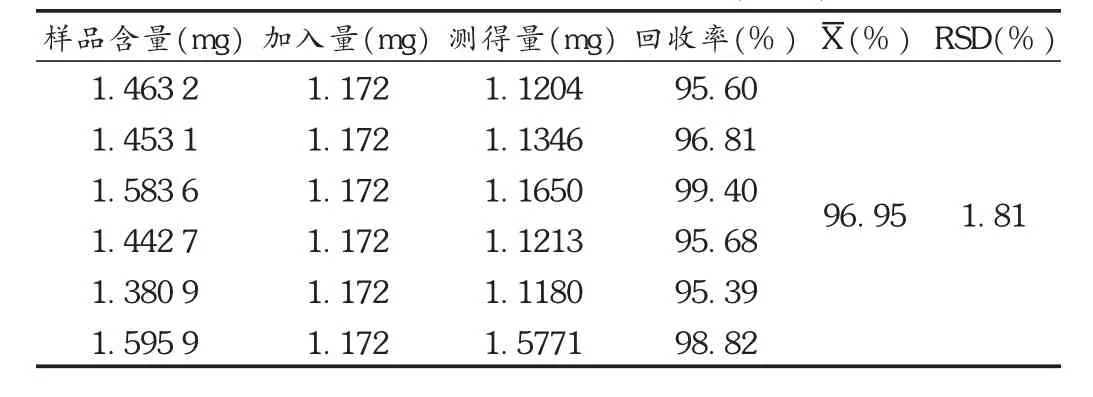
表1 芍药苷加样回收试验结果(n=6)
2.2.4 样品含量测定
分别精密吸取对照品溶液和供试品溶液各10 μL,进样,测定3批次样品中芍药苷的含量。结果批号为20130404,20130405,20130406的 3批次样品中芍药苷的平均含量分别为 32.53,33.19,32.94 mg /g。
3 讨论
本品具有健脾益肾、活血通络之功效,用于治疗糖尿病周围神经病变。赤芍为方中要药,具有活血通经之功效,有效成分芍药苷具有扩张血管、镇静、镇痛、抗炎等作用。故选择芍药苷作为本品的含量测定指标,与本品的功能具有较高的相关性。
用50%乙醇为提取溶剂,对回流法和超声法处理样品进行比较,结果表明,超声法提取效率较高,故选用超声法提取芍药苷。用 50% 乙醇为提取溶剂,分别采用超声 10,20,30,40,60 min进行提取。结果表明,超声40 min可以提取完全,故选择40 min为样品提取时间。
流动相选择中,分别考察了乙腈-0.1%磷酸溶液[1]、甲醇-水等系统[2-4]的不同配比。结果表明,甲醇-0.05 mol/L磷酸二氢钾(29∶71)为流动相最为理想,此条件下芍药苷与其他成分能够较好地分离,阴性无干扰,且样品处理方法简单,可作为本品质量控制的理想方法。
参考文献:
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:96-97.
[2]梁 帅,王路宏,李 岩.高效液相色谱法测定复光颗粒中芍药苷含量[J].中国药业,2012,21(15):40-41.
[3]李玉英,吴俊贤,郑 惠,等.高效液相色谱法测定尪痹片中芍药苷含量[J].中国药业,2013,22(11):43-44.
[4]邓双炳,石 岩,胡玉花,等.HPLC测定脾肾两助丸中芍药苷的含量[J].中国实验方剂学杂志,2013,19(5):123-125.
猜你喜欢
今日农业(2022年13期)2022-09-15
新作文·小学低年级版(2022年3期)2022-08-30
金沙江文艺(2022年1期)2022-02-04
上海文化(文化研究)(2021年6期)2022-01-07
智慧少年·故事叮当(2021年5期)2021-08-23
化学教与学(2021年12期)2021-02-18
中学生数理化(高中版.高考理化)(2020年1期)2020-11-24
中成药(2019年12期)2020-01-04
时代邮刊·下半月(2019年8期)2019-09-10
今日农业(2019年15期)2019-01-03
