
美洛昔康口腔崩解片中美洛昔康含量测定方法研究
2014-05-03 06:15丁捷飞冯定军胡黎凤
中国药业 2014年5期
丁捷飞,冯定军,胡黎凤,俞 伟
(宁波立华制药有限公司,浙江 宁波 315174)
美洛昔康(meloxicam)为新型非甾体抗炎药(NSAIDs),临床主要用于风湿性关节炎、类风湿性关节炎等各类炎症及疼痛的治疗,止痛作用较强,同时可大幅度降低非甾体抗炎药的胃肠和肾脏不良反应,尤其能降低肠胃穿孔、溃疡和出血的概率[1-2]。有关美洛昔康制剂的含量测定方法已有文献报道[3-4],但关于美洛昔康口腔崩解片中美洛昔康的含量测定尚未见文献报道。为此,本试验中采用简捷、方便的前处理方法,以反相高效液相色谱(RP-HPLC)法测定美洛昔康口腔崩解片中美洛昔康的含量,并对测定方法进行了方法学考察。现报道如下。
1 仪器与试药
Agilent HP1100型高效液相色谱仪(四元泵VWD检测器,真空脱气机);梅特勒AE240型电子分析天平。甲醇(色谱纯,上海陆忠化学试剂厂);醋酸铵(分析纯,宜兴市化学试剂三厂);美洛昔康对照品(中国药品生物制品检定所,批号为100679-200401,供含量测定用);美洛昔康口腔崩解片(宁波立华制药有限公司,批号分别为 12091201,12091202,12091203,规格为每片含美洛昔康 7.5 mg)。
2 方法与结果
2.1 色谱条件
色谱柱:Kromasil C18柱(250 mm ×4.6 mm,5 μm);柱温:室温;流动相:0.1 mol/L 醋酸铵溶液 - 甲醇(50 ∶50);流速:1.0 mL /min;检测波长:271 nm。
2.2 溶液制备
精密称取美洛昔康对照品10 mg,置100 mL容量瓶中,加入适量流动相,振摇使溶解,超声助溶10 min,静置待溶液恢复至室温,用流动相稀释至刻度,摇匀;精密量取5 mL,置10 mL容量瓶中,用流动相稀释至刻度,作为对照品溶液。取样品20片,精密称定,研细,精密称取本品粉末适量(约含美洛昔康10 mg),置100 mL容量瓶中,加流动相适量,超声助溶使充分溶解(约10 min),冷却至室温,用流动相稀释至刻度,滤过,精密量取续滤液5 mL,置10 mL容量瓶中,用流动相稀释至刻度,作为供试品溶液。
2.3 方法学考察
系统适用性试验:取对照品溶液及供试品溶液各20 μL,分别进样测定。结果在较短时间内被测组分完全洗脱,理论板数为8 300,拖尾因子为1.03,对照品溶液和供试品溶液在拟订色谱条件下均有较好的分离效果,见图1。
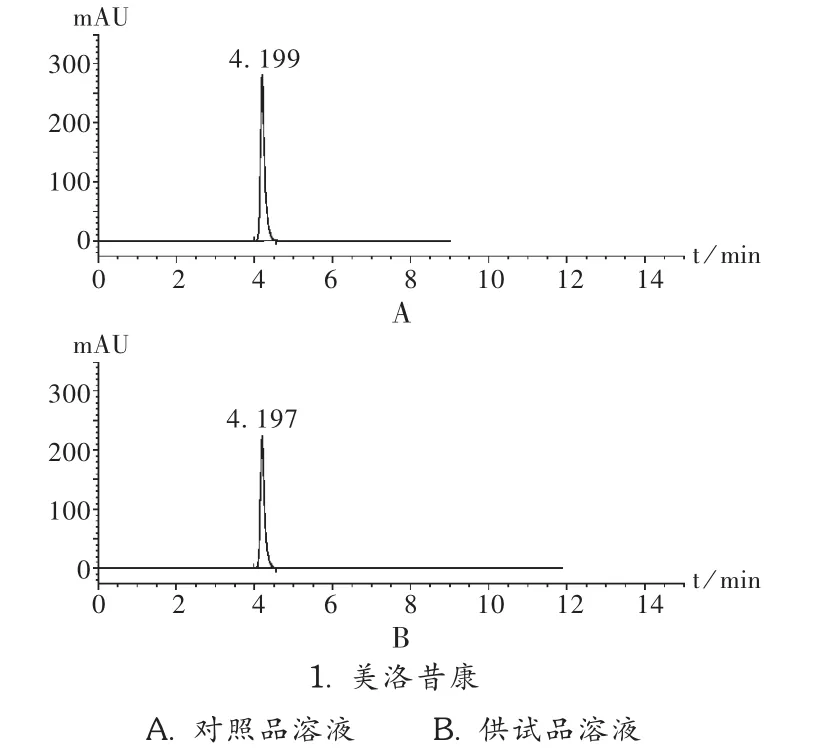
图1 高效液相色谱图
专属性试验[5]:按处方量分别精密称取全辅料、原料药、试片各适量,用流动相稀释成含主成分质量浓度为100 μg/mL的全辅料贮备液、原料药贮备液、试片贮备液(其中全辅料、试片需滤过,取续滤液作为贮备液)。精密量取以上贮备液各5 mL,置10 mL容量瓶中,用流动相稀释至刻度,分别制得全辅料、原料药及试片的供试品溶液。按拟订色谱条件分别进样测定,记录色谱图。结果溶剂、全辅料、原料药、试片的杂峰个数分别为 1,3,1,3个,且美洛昔康主峰前后无其他杂质峰干扰,溶剂峰和全辅料峰也都不干扰主峰的测定。
线性关系考察:分别精密量取对照品溶液 4.0,4.5,5.0,5.5,6.0 mL,置 10 mL 容量瓶中,用流动相稀释至刻度。按拟订色谱条件,分别进样,记录色谱图,以其峰面积与相对应的质量浓度作线性回归,得回归方程 Y=34 321X+19 501,r=0.999 8(n=5)。结果表明,美洛昔康质量浓度在40~60 μg/mL范围内与峰面积呈良好线性关系。
重复性试验:精密称取样品适量(相当于含美洛昔康10 mg),依法制备供试品溶液,按拟订色谱条件分别进样测定6次。结果平均峰面积为 1 763 034,RSD =0.22%(n=6),表明方法重复性较好。
稳定性试验:精密吸取同一供试品溶液,分别于 0,1,2,4,8,24 h时进样。结果平均峰面积为 1 764 637,1 765 144,1 763 436,1 763 224,1 764 718,1 761 098,RSD =0.17% (n=6),表明供试品溶液室温下放置24 h以内有较好的稳定性。
回收率试验:精密称 取 1.33倍处方量80%,100%,120%的原料药,分别置100 mL容量瓶中,同时按处方加入适量的全辅料,依法制备供试品溶液,按拟订色谱条件平行测定3次,计算回收率。结果见表1。
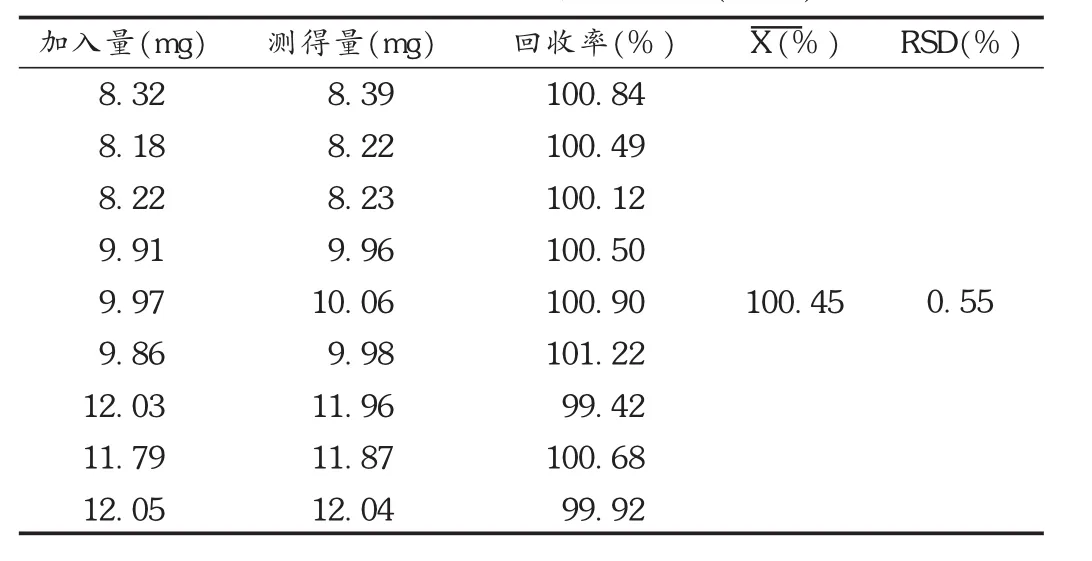
表1 美洛昔康回收率试验结果(n=9)
2.4 样品含量测定
取对照品溶液和供试品溶液,按拟订色谱条件进样,按外标法以峰面积计算美洛昔康含量。结果批号为12091201,12091202,12091203 的 3 批样品中每片含美洛昔康 7.5,7.6,7.5 mg。
3 讨论
按照产品处方,分别用水配制质量浓度(按美洛昔康计)为50 μg/mL的全辅料、原料药及试片的供试品溶液,在200~400 nm波长内进行紫外扫描。结果表明,全辅料在271 nm波长处有较大的紫外吸收,干扰主成分的分析,不能采用紫外分光光度法进行含量分析。原料药及试片在271 nm波长处均有最大吸收,故确定本品的检测波长为271 nm。
参考2010年版《中国药典(二部)》美洛昔康有关物质测定方法[6-7],精密称取本品适量,用流动相制成质量浓度为1 g/L的供试品溶液,进样20 μL,记录色谱图。结果表明,该色谱条件下美洛昔康主峰保留时间为17.90 min,保留时间太长。调整流动相比例为 0.1 mol/L 醋酸铵溶液 - 甲醇(50 ∶50),将上述供试品溶液20 μL注入高效液相色谱仪,记录色谱图。结果表明,调整比例后美洛昔康主峰保留时间为4.2 min,保留时间适宜,可以对该色谱条件进行进一步研究。
2010年版《中国药典(二部)》采用滴定法测定美洛昔康原料药的含量,在当前国家药品质量标准日趋严格的情况下,这种分析方法将逐渐为高效液相色谱法所取代。本试验结果可对美洛昔康原料药及其制剂的质量标准研究起到参考作用。
参考文献:
[1]鲁统栋,张云良,程维国.非甾体抗炎新药美洛昔康[J].中国药业,1999,8(12):56.
[2]Xu XC.COX-2 inhibitors in cancer treatment and prevention,a recent development[J].Anticancer Drugs,2002,13:127-137.
[3]冯新生,冯巧枝,刘富民.高效液相色谱法测定美洛昔康胶囊的含量[J].中国医院药学杂志,2003,23(4):219-221.
[4]陈 雯.HPLC法测定美洛昔康分散片的含量[J].中国药师,2009,12(6):765-766.
[5]杨仲文,沈玲佳,刘 红.美洛昔康中有关物质的HPLC测定[J].中国医药工业杂志,2003,34(11) :575-576.
[6]杨岫岩,董 怡.非甾体类抗炎药与环氧化酶异构体[J].中华内科杂志,1996,35(12):834-835.
[7]国家药典委员会.中华人民共和国药典(二部)[M].北京:中国医药科技出版社,2010:448.
猜你喜欢
腐蚀与防护(2021年8期)2021-09-07
石油学报(石油加工)(2021年3期)2021-05-14
热力发电(2020年9期)2020-12-05
哈尔滨轴承(2020年1期)2020-11-03
纺织服装流行趋势展望(2020年1期)2020-02-01
传媒评论(2019年6期)2019-10-14
中国经济信息(2017年17期)2017-09-09
纺织服装流行趋势展望(2016年6期)2016-05-04
纺织服装流行趋势展望(2016年4期)2016-05-04
纺织服装流行趋势展望(2016年1期)2016-05-04
